Article Text

Statistics from Altmetric.com
1. FOREWORD
I am delighted to write the foreword for this consensus statement which updates the “Recommendations on the management of pulmonary hypertension in clinical practice” of 2001. The consensus statement reflects contemporary practice in the management of this uncommon, deadly but now treatable condition in the pulmonary hypertension designated centres in the UK and Ireland. It matches and complements international guidelines, which are currently under revision.
In addition to health professionals who encounter affected patients within national centres or in other fields of practice, the statement is a comprehensive but readily accessible source of information for commissioners and managers of specialised services.
Initially the evidence on which to base the care of patients with rare and deadly diseases often rests on the experience and judgement of those who deliver daily care, the collection of clinical, epidemiological and pathological data, and the assiduous construction of informative registers. This familiar discipline has enabled the advances summarised in this document.
The first challenge to health service commissioners is to ensure that all patients with pulmonary hypertension have access to appropriate therapy as quickly as possible. Delay in making the diagnosis has the same consequences as delay in those with cancer. Regrettably “postcode prescribing” and its consequences still persist.
As for other uncommon conditions, continued progress in developing effective therapies rests on specialised centres working in concert to develop and participate in well-designed clinical trials, particularly trials of combination therapy.
This statement is a testament to the major advances in therapy made over the last 10 years, progress those of us who have cared for such patients over more than 30 years could not have envisaged.
Professor Dame Carol M Black DBE, MD, FRCP, MACP, FMEDSCT
Lead clinicians
Gerry Coghlan, Royal Free Hospital, London, UK
Paul A Corris (Co-editor), Freeman Hospital, Newcastle, UK
Sean Gaine, Mater Misericordae, Dublin, Ireland
Michael A Gatzoulis, Royal Brompton Hospital, London, UK
J Simon R Gibbs (Chairman and co-editor), Hammersmith Hospital, London, UK
Sheila G Haworth, Great Ormond Street Hospital for Children, London, UK
David G Kiely, Royal Hallamshire Hospital, Sheffield, UK
Andrew Peacock, Western Infirmary, Glasgow, UK
Joanna Pepke-Zaba, Papworth Hospital, Papworth Everard, UK
Pulmonary hypertension clinicians
Carol Black, Royal Free Hospital, London, UK
Charlie Elliot, Royal Hallamshire Hospital, Sheffield, UK
Andrew J Fisher, Freeman Hospital, Newcastle UK
Clive Handler, Royal Free Hospital, London, UK
Luke Howard, Hammersmith Hospital, London, UK
Rodney Hughes, Royal Hallamshire Hospital, Sheffield, UK
David P Jenkins, Papworth Hospital, Papworth Everard, UK
Martin Johnson, Western Infirmary, Glasgow, UK
Jim Lordan, Freeman Hospital, Newcastle, UK
Guy MacGowan, Freeman Hospital, Newcastle UK
Nick Morrell, Addenbrookes Hospital, Cambridge, UK
Ingram Schulze-Neick, Great Ormond Street Hospital for Children, London, UK
Karen Sheares, Papworth Hospital, Papworth Everard, UK
Martin Wilkins, Hammersmith Hospital, London, UK
John Wort, Royal Brompton Hospital, London, UK
Pulmonary hypertension clinical nurse specialists
Agnes Crozier, Western Infirmary, Glasgow, UK
Clare Das, Royal Free Hospital, London, UK
Julia De Soyza, Freeman Hospital, Newcastle, UK
Sinead Doherty, Mater Misericordae, Dublin, Ireland
Yvette Flynn, Great Ormond Street Hospital for Children, London, UK
Wendy Gin-Sing, Hammersmith Hospital, London, UK
Carl Harries, Royal Brompton Hospital, London, UK
Maureen Rootes, Papworth Hospital, Papworth Everard, UK
Invited individuals and organisations
Geoffey Carroll, Medical Director, Health Commission Wales
Pulmonary Hypertension Association (UK)
Iain Armstrong, Royal Hallamshire Hospital, Sheffield, UK
British Congenital Cardiac Association
John Gibbs, Leeds General Infirmary Leeds, UK
British Society of Human Genetics
Richard Trembath, Guy’s Hospital, London, UK
2. INTRODUCTION
In 2001 the BCS Guidelines and Medical Practice Committee published their “Recommendations on the management of pulmonary hypertension in clinical practice”1 which was approved by the British Thoracic Society (BTS) and the British Society of Rheumatology (BSR).
Subsequently the National Pulmonary Hypertension Centres of the UK and Ireland Physicians Committee was constituted to represent all of the designated centres. This statement is issued by this Committee and represents the views of healthcare professionals who provide expert management of pulmonary hypertension (PH) in designated centres.
Major advances in the clinical management of PH have been made in the time which has elapsed since the 2001 publication. The purpose of this consensus statement is to update the 2001 recommendations to reflect contemporary clinical practice in designated centres routinely managing PH in the UK and Ireland. It is published to inform other health care professionals, commissioners and managers who are responsible for delivering healthcare.
Since 2001 formal guidelines for the management of pulmonary arterial hypertension (PAH) have been published by the European Society of Cardiology (ESC)2 and the American College of Chest Physicians (ACCP).3–6 Both of these guidelines are in the process of being updated for publication in 2008–2009. Furthermore the clinical nomenclature was revised7 and clinical practice recommendations made8–10 at the 3rd World Symposium on Pulmonary Arterial Hypertension in 2004. These will also be updated in 2008 at the 4th World Symposium.
We have not sought to replicate international guidelines and thus there is no grading of evidence or recommendations. Instead this consensus statement is intended to complement these PAH guidelines with specific emphasis on UK and Irish practice, as well as to extend them to other forms of PH. We recognise that in such a rapidly advancing field of clinical practice there will be a need to revise this statement in due course.
2.1 Evolution of treatment
Patients with PAH who do not receive disease-targeted therapy have a poor quality of life (QoL) and high mortality at rates similar to many cancers. In 1996 the first randomised trial of drug therapy in PAH demonstrated benefit with epoprostenol, establishing this as therapy for severe idiopathic pulmonary arterial hypertension (IPAH) in World Health Organization (WHO) functional classes III and IV.11
Over the last 10 years randomised, placebo controlled trials of other prostacyclin analogues, endothelin receptor antagonists and phosphodiesterase inhibitors have shown significant benefit to patients with PAH, with improved survival and functional class.
In the UK designated centres, the number of patients on these treatments in both clinical practice and clinical trials on 31 March was 638 in 2004, 912 in 2005, 1242 in 2006 and 1499 in 2007. This represents a total of 24.9 patients treated per million population based on the size of the UK population in mid 2005.12 It is expected that this number will increase as patients survive longer, more patients come to medical attention and the indications for disease-targeted therapy expands.
Abbreviations
ACCP: American College of Chest Physicians
ALK-1: activin receptor-like kinase 1
APAH: associated pulmonary arterial hypertension
ATS: American Thoracic Society
BCS: British Cardiovascular Society
BLT: bilateral lung transplantation
BMPRII: bone morphogenetic protein receptor type II
BNP: brain natriuretic peptide
BSR: British Society of Rheumatology
BTS: British Thoracic Society
cAMP: cyclic adenosine monophosphate
CAMPHOR: Cambridge Pulmonary Hypertension Outcome Review
CCAD: Central Cardiac Audit Database
cGMP: cyclic guanosine monophosphate
COPD: chronic obstructive pulmonary disease
CPET: cardiopulmonary exercise test
CT: computed tomography scan
CTD: connective tissue disease
CTEPH: chronic thromboembolic pulmonary hypertension
DLCO: lung diffusing capacity
ECG: electrocardiogram
ERA: endothelin receptor antagonist
ETA: endothelin A
ETB: endothelin B
ESC: European Society of Cardiology
FPAH: familial pulmonary arterial hypertension
GOSHC: Great Ormond Street Hospital for Children
GP: general practitioner
GUCH: grown-up congenital heart disease
HIV: human immunodeficiency virus
HRQoL: health-related quality of life
ILD: interstitial lung disease
INR: international normalised ratio
IPAH: idiopathic pulmonary arterial hypertension
ISHLT: International Society of Heart and Lung Transplantation
IVC: inferior vena cava
LTOT: long term oxygen therapy
MCTD: mixed connective tissue disease
MR: magnetic resonance
NCG: National Commissioning Group (formerly NSCAG)
NHS: National Health Service
NO: nitric oxide
NSCAG: National Specialist Commissioning Advisory Group
NSD: National Service Division
NYHA: New York Heart Association
PAH: pulmonary arterial hypertension
PAP: pulmonary arterial pressure
PASP: pulmonary arterial systolic pressure (estimated by echocardiography)
PCH: pulmonary capillary haemangiomatosis
PCT: Primary Care Trust
PCWP: pulmonary capillary wedge pressure
PDE: phosphodiesterase
PEA: pulmonary endarterectomy
PFO: patent foramen ovale
PH: pulmonary hypertension
Pro-NT BNP: pro-N terminal brain natriuretic peptide
PVOD: pulmonary veno-occlusive disease
PVR: pulmonary vascular resistance
QoL: quality of life
SCG: Specialist Commissioning Group
SLE: systemic lupus erythematosis
SLT: single lung transplantation
SSc: scleroderma
TGFβ: transforming growth factor β
TR: tricuspid regurgitation
VIP: vasoactive intestinal polypeptide
WHO: World Health Organization
2.2 Centres designated to manage pulmonary hypertension
The purpose of designated centres is to provide best clinical practice, well coordinated patient care, clinical research, and advice for those who are managing patients but are not specialists in PH. Care is provided by multiprofessional teams for inpatients, day cases and outpatients with 24 h cover.
Centres designated to manage PH are shown in table 1. There are seven hospitals in England, one in Scotland and one in Ireland. Wales and Northern Ireland refer patients to UK centres and may develop their own or satellite centres in the future. This directory of centres is kept current at www.thephdirectory.com.
Formal designation of centres was undertaken by the National Specialist Commissioning Advisory Group (NSCAG) of the Department of Health in England in 2001, the National Service Division (NSD) of the Scottish Parliament in Scotland in 1998, and the Health Service Executive in Ireland. NSCAG was replaced by the National Commissioning Group (NCG) in 2007. These centres are monitored by their designating bodies by regular site visits and audit against agreed Standards of Care. Audit data will become centralised in the National Health Service (NHS) in 2008 when it is collected from all designated centres by the Central Cardiac Audit Database (CCAD).
2.3 Commissioning of pulmonary hypertension in England
In England, PH is included within the list of defined specialist services issued by the Department of Health. For adults, this means that Primary Care Trusts (PCTs) are required to commission the service through formal collaborative arrangements established by the Specialist Commissioning Group (SCG) responsible for their area.
At national level specialist commissioners are working with the six designated adult centre lead clinicians to formulate service development strategies and policies aimed at ensuring a consistent and dynamic approach in the future.
Funding of expensive disease-targeted therapies is provided on an individual patient basis by PCTs to whom applications must be made for each patient by a designated centre. Some PCTs have formed consortia to fund the cost of treatment according to strict criteria without the need for individual applications.
For children, NCG not only designates but funds the service and drug therapies. The Pulmonary Endarterectomy (PEA) Service is separately NCG designated and centrally funded.
2.4 Commissioning of pulmonary hypertension in Scotland
The Scottish Pulmonary Vascular Unit is commissioned to provide services for Scotland and is centrally funded by the NSD.
2.5 Commissioning of pulmonary hypertension in Ireland
A single PH Unit has been commissioned to provide a service for the whole of the Republic of Ireland and is centrally funded on a yearly basis by the Health Service Executive.
2.6 Collection of audit data
Currently all designated centres collect audit data in local databases. In 2008 the UK data will be centralised in the CCAD, part of the National Clinical Audit Support Programme of the NHS.
3. NOMENCLATURE
3.1 Clinical classification
The clinical classification of PH is key to making an accurate diagnosis and guides treatment. It was updated in 20047 (box 1).
Box 1: Revised clinical classification of pulmonary hypertension (Venice 2003) following the previous Evian classification (described in the 2001 BCS recommendations)
1. Pulmonary arterial hypertension (PAH)
1.1. Idiopathic (IPAH)
1.2. Familial (FPAH)
1.3. Associated with (APAH):
1.3.1. Collagen vascular disease
1.3.2. Congenital systemic-to-pulmonary shunts
1.3.3. Portal hypertension
1.3.4. HIV infection
1.3.5. Drugs and toxins
1.3.6. Other (thyroid disorders, glycogen storage disease, Gaucher’s disease, hereditary haemorrhagic telangiectasia, haemoglobinopathies, myeloproliferative disorders, splenectomy)
1.4. Associated with significant venous or capillary involvement
1.4.1. Pulmonary veno-occlusive disease (PVOD)
1.4.2. Pulmonary capillary haemangiomatosis (PCH)
1.5. Persistent pulmonary hypertension of the newborn
2. Pulmonary hypertension with left heart disease
2.1. Left-sided atrial or ventricular heart disease
2.2. Left-sided valvular heart disease
3. Pulmonary hypertension associated with lung diseases and/or hypoxaemia
3.1. Chronic obstructive pulmonary disease
3.2. Interstitial lung disease
3.3. Sleep disordered breathing
3.4. Alveolar hypoventilation disorders
3.5. Chronic exposure to high altitude
3.6. Developmental abnormalities
4. Pulmonary hypertension due to chronic thrombotic and/or embolic disease
4.1. Thromboembolic obstruction of proximal pulmonary arteries
4.2. Thromboembolic obstruction of distal pulmonary arteries
4.3. Non-thrombotic pulmonary embolism (tumour, parasites, foreign material)
5. Miscellaneous
Sarcoidosis, histiocytosis X, lymphangiomatosis, compression of pulmonary vessels (adenopathy, tumour, fibrosing mediastinitis)
The classification is based upon groups of diseases causing PH which demonstrate similarities in clinical presentation, pathophysiology and therapeutic options.
The broad influence of the clinical classification on management is seen in table 2.
3.2 Functional class
The severity of PH is assessed according to a modification of the New York Heart Association (NYHA) functional classification13 shown in box 2. It has long been recognised that symptomatic severity is related to prognosis14 and this remains so in contemporary practice (fig 1)15 emphasising the need for early referral for investigation and treatment.

Box 2: Functional classification of pulmonary hypertension modified after the New York Heart Association functional classification according to the World Health Organization 1998
Class I: Patients with pulmonary hypertension but without resulting limitation of physical activity. Ordinary physical activity does not cause undue dyspnoea or fatigue, chest pain or near syncope.
Class II: Patients with pulmonary hypertension resulting in slight limitation of physical activity. They are comfortable at rest. Ordinary physical activity causes undue dyspnoea or fatigue, chest pain or near syncope.
Class III: Patients with pulmonary hypertension resulting in marked limitation of physical activity. They are comfortable at rest. Less than ordinary activity causes undue dyspnoea or fatigue, chest pain or near syncope.
Class IV: Patients with pulmonary hypertension with inability to carry out any physical activity without symptoms. These patients manifest signs of right heart failure. Dyspnoea and/or fatigue may even be present at rest. Discomfort is increased by any physical activity.
4. PATHOPHYSIOLOGY AND GENETICS OF PULMONARY ARTERIAL HYPERTENSION: LINKS TO TREATMENTS
The pathology of PAH is characterised by luminal obliteration of small pulmonary arteries. This process of vascular remodelling involves proliferation of smooth muscle cells, fibroblasts and endothelial cells in the vessel wall.16–18 In severe forms of PH, the formation of a neointima is observed forming concentric intimal lesions. Abnormal endothelial cell proliferation results in the formation of plexiform lesions (fig 2). The most severe forms of precapillary PH are usually pathologically indistinguishable.

A number of mediators and growth factors have been shown to be involved in driving the cellular changes. Increased circulating and local expression of endothelin-1 is observed in patients with PAH.19 ,20 As well as being a potent vasoconstrictor, endothelin stimulates smooth muscle and fibroblast proliferation via the endothelin A (ETA) and/or endothelin B (ETB) receptors, which are increased in small hypertensive pulmonary arteries.21 Circulating levels of serotonin are also elevated in PAH.22 Serotonin stimulates mitogenesis of vascular cells via serotonin receptors, including 5HT2A, 5HT2B and 5HT1B.18 In human pulmonary artery smooth muscle cells, a major proliferative pathway involves activation of mitogen activated protein kinases via the serotonin transporter.23 Increased expression of the transporter is found in hypertensive arteries.
A relative deficiency of vasodilator pathways is observed in severe PAH, an imbalance which enhances the activity of mitogenic and vasoconstrictor pathways. Patients with PAH produce less endothelial-derived prostacyclin, and have reduced expression of nitric oxide (NO) synthase and vasoconstrictive thromboxane.24 More recent studies have also shown a deficiency of the neuropeptide vasodilator vasoactive intestinal polypeptide (VIP) in the lungs of patients with PAH.25 Many of the important vasodilator pathways also exert antiproliferative effects on vascular cells. The deficiency of these key vasodilator pathways has provided the rationale for therapies.
Other important pathways involved in the process of pulmonary vascular remodelling include changes in potassium channel (Kv1.5 and 2.1) expression, activation of vascular elastases, and increased expression of inflammatory cytokines and chemokines.18
The genetics of PAH is described in section 5.3.1.
5. OBJECTIVES AND PRIORITIES FOR INVESTIGATION
5.1 Definition of pulmonary hypertension
PH is defined as a mean pulmonary arterial pressure (PAP) of >25 mm Hg at rest or >30 mm Hg on exercise at cardiac catheterisation.26 PAH also requires a pulmonary capillary wedge pressure (PCWP) ⩽15 mm Hg and a pulmonary vascular resistance (PVR) ⩾240 dynes/s/cm5.2
5.2 When to suspect pulmonary hypertension
The principal symptoms of PH are non-specific and the clinical signs subtle until patients present with advanced disease.26 As a consequence the diagnosis is most readily made where a systematic approach is taken to investigation, and high risk patients are targeted with screening programmes.
Clinical suspicion should arise in any patient presenting with breathlessness without overt signs of specific heart or pulmonary disease, particularly in diseases which may be associated with PH (box 1). While breathlessness is the most common symptom, patients may also present with chest pain, syncope, fatigue, weakness and abdominal distension.26 Frequently there is a delay of up to 3 years between first symptom and diagnosis and this interval has remained the same over the last 10 years.27
The precordial signs of PH include right ventricular lift, accentuated pulmonary component of the second heart sound, a pansystolic murmur of tricuspid regurgitation, a diastolic murmur of pulmonary regurgitation and a right ventricular third sound. Jugular venous distension, hepatomegaly, peripheral oedema, ascites and cold extremities characterise patients in a more advanced state with right ventricular failure at rest; central cyanosis may also be present. Ankle swelling occurs late in the natural history of the disease.
When faced with breathlessness of unknown cause, spirometry is a useful screening test to exclude common respiratory disease. A chest x ray and ECG should be performed since these tests are abnormal in 80–90% of patients presenting with symptoms caused by established PH.1 The ECG may demonstrate right ventricular hypertrophy and strain and right atrial dilatation. The ECG alone has inadequate sensitivity (55%) and specificity (70%) to be a screening tool for detecting PH.28
Doppler echocardiography is the most useful non-invasive investigation and allows an estimate of pulmonary arterial systolic pressure (PASP).29 Its sensitivity and specificity in identifying PH depends on the population and limitations relate primarily to technical aspects of this technique. The estimated upper limit for PASP in 95% of normal subjects is 37.2 mm Hg.30 A PASP >40 mm Hg was found in 6% of those >50 years old and 5% of those with a body mass index (BMI) >30 kg/m. Twenty-eight per cent of normal subjects have a PASP >30 mm Hg, and the expected upper limit of PASP may be as high as 40 mm Hg in older or obese subjects. Mild PH has been defined as a peak tricuspid regurgitation (TR) velocity of 2.8–3.4 m/s with a normal right atrial pressure.2
Computed tomographic (CT) scanning also provides useful information on right ventricular and pulmonary artery size, raising the possibility of PH which had not otherwise been suspected.
RECOMMENDATIONS
1. PH should be considered in all patients presenting with breathlessness in the absence of an alternative cause of cardiorespiratory disease. The presence of progressive breathlessness associated with chest pain or syncope should particularly alert the clinician to this diagnosis.
2. While ECG and chest x ray are often abnormal at presentation, the sensitivity of these investigations is such that normal appearances do not exclude PH.
3. Spirometry should be performed to detect common respiratory diseases.
4. Doppler echocardiography is the best screening investigation for PH.
5.3 Screening at risk populations for pulmonary hypertension
5.3.1 Genetic screening for pulmonary arterial hypertension
Incident data from specialist centres indicate the minimum frequency of recognised familial pulmonary arterial hypertension (FPAH) as 5–10% of referrals. Risk for FPAH is conferred by a heterozygous loss of function mutation of receptor members of the TGFβ superfamily, most commonly defects in the gene encoding the type II receptor, BMPRII.31 ,32 Rarely, mutations of the type I receptor, ALK-l, have been detected in PAH subjects who may also exhibit the clinical features characteristic of hereditary haemorrhagic telangiectasia.33 A significant proportion of sporadic cases (classified as IPAH) have germline TGFβ mutations34 while the frequency of detectable mutations appears lower in childhood onset disease.35 Incidence and prevalence data for populations for FPAH/IPAH have not been reported.
An evidence base for the management of “at risk” individuals for PAH requires further research and will require modification should disease prevention or modification strategies emerge. The provision of information regarding risk and opportunities for risk resolution in monogenic disorders is a recognised function of genetic services, which should be extended to FPAH kindreds.
Indications for clinical and molecular genetic screening in IPAH remain unclear and require further research. Empirical data suggest low disease risks to first and second degree relatives in IPAH. Presentation in childhood raises specific parental concern for occurrence of disease in siblings (recurrence risk) and/or the unborn child (offspring risk) which may require referral for specialist genetic counselling. Mutation analysis has provided no insight into the clinical variability of PAH, particularly age of onset which may vary significantly within families.35 ,36 The genetic basis of associated forms of PAH remains unclear.
RECOMMENDATIONS
5. Recognised familial cases of PAH should be offered family-based risk assessment including genetic counselling.
6. Molecular genetic testing may be indicated in FPAH following comprehensive genetic counselling for (a) resolution of individual risk and (b) family planning. Joint management of “at risk” individuals within recognised families between genetic services and specialist PAH centres is indicated. Clinical monitoring of at risk relatives is indicated for early detection of disease and management of symptoms. The frequency at which this should be conducted is unknown.
7. Close (first degree) relatives of index IPAH patients should be provided with written information of the genetic basis of the disorder, including recognition of the low (<5%) recurrence and offspring risk. The role of clinical monitoring in this group is unknown.
8. Parental anxiety regarding recurrence and/or offspring risk following childhood onset presentation with IPAH is an indication for genetic services referral with provision for clinical assessment of “at risk” family members.
5.3.2 Associated pulmonary arterial hypertension
PAH is commonly seen in association with connective tissue disease (CTD),37 congenital heart disease,38 sickle cell disease,39 portal hypertension40 and HIV infection.41 This has resulted in a number of screening regimens to identify PAH in at risk groups ranging from investigating patients with symptoms of breathlessness to interval screening of asymptomatic individuals.
RECOMMENDATIONS
9. Screening of breathless patients should be performed in diseases where PAH is a known complication. Right heart catheterisation should be undertaken when Doppler echocardiography measures a peak TR velocity of ⩾2.8 m/s with a normal right atrial pressure (equivalent to 36 mm Hg).
5.3.2.1 Connective tissue disease
Pulmonary hypertension is a well known complication of CTD, particularly in limited cutaneous scleroderma (SSc) where the prevalence in this group is 12%,42 and mixed CTD (MCTD) with U1 RNP antibodies.43 PAH is recognised by an increased Doppler peak TR velocity at echocardiography and reduced lung diffusing capacity (DLCO).37 ,44 ,45
RECOMMENDATIONS
10. Screening should be performed annually in patients with limited cutaneous SSc or MCTD with U1 RNP antibodies, using echocardiography and DLCO. Right heart catheterisation should be performed in all cases with a peak TR velocity of ⩾2.8 m/s on echocardiography or a reduction in DLCO of 50% in the absence of interstitial lung disease (ILD). Patients with other CTDs are screened only in the presence of symptoms.
5.3.2.2 Porto-pulmonary hypertension
The prevalence of PAH in patients undergoing liver transplantation is 4.0–3.5%.46 In addition porto-systemic shunts increase the risk of developing PAH.47
RECOMMENDATIONS
11. All patients with portal hypertension and cirrhosis should undergo echocardiography if liver transplantation is planned.
5.3.2.3 Haemolytic anaemia
PAH is increasingly recognised in congenital haemolytic anaemias including sickle cell disease39 and thalassaemia.48 ,49 It is not yet clear whether these patients benefit from PAH disease-targeted therapies.
RECOMMENDATIONS
12. Screening for PAH is not routine in patients with haemolytic anaemia.
5.3.2.4. HIV infection
PAH is a rare complication of HIV with a cumulative incidence of 0.57% on an annual incidence of 0.1%.41
RECOMMENDATIONS
13. Screening for PAH is not routine in patients with HIV infection.
5.3.3. Pulmonary embolism
Chronic thromboembolic pulmonary hypertension (CTEPH) is a complication of venous thromboembolism. Up to 4% of patients with idiopathic pulmonary embolism may develop CTEPH.50 Patients at greatest risk include those with previous episodes of venous thromboembolism, massive and sub-massive pulmonary embolism,51 an elevated PASP on admission or elevated pressure 2 months following initial presentation.
RECOMMENDATIONS
14. Patients with previous venous thromboembolism who are breathless should undergo echocardiography. Patients with massive or sub-massive pulmonary thromboembolism should undergo echocardiography 6–12 weeks following the index event. Where echocardiography is inconclusive and symptoms persist, consider contrast CT thorax.
5.4 Criteria for referral to pulmonary hypertension centres
Referrals are accepted at designated centres where screening investigations suggest PH for which there is not a cardiac or respiratory cause. Right heart catheterisation is not encouraged before referral unless individual cases are discussed with a designated centre, and the referring physician routinely undertakes right heart catheterisation with vasodilator studies.
RECOMMENDATIONS
15. Adults with confirmed or suspected PAH, CTEPH, a miscellaneous cause of PH, or where the cause of PH is unclear should be referred to a designated centre. Referral should be considered in cases of PH in hypoxic lung disease or cardiac disease, but only if symptoms or estimated PASP at echocardiography seems excessive (>60 mm Hg) or the patient has another disease which may be associated with PAH.
16. Children should be referred to the UK Children’s Service if they have confirmed or suspected IPAH or FPAH. The UK Children’s Service will also accept referral of and/or be available to give advice for all children with persistent neonatal PH beyond the first month of life, sustained, postoperative PH, inoperable congenital heart disease with PH, parenchymal lung disorders/disease with PH, miscellaneous causes of PH, or PH of uncertain cause.
17. All patients should have an ECG, chest x ray, transthoracic echocardiogram, and spirometry in adults. If possible, all patients should be seen by a consultant in cardiology or respiratory medicine before referral to a designated centre.
18. Patients with PH may deteriorate rapidly. It is important that referrals are not delayed in order to undertake more extensive investigation if it is clear that PH is the dominant problem.
5.5 Investigation at pulmonary hypertension centres
The purpose of investigation is to confirm or exclude the diagnosis of PH, and if present to determine the aetiology and severity of PH (table 3 and box 3).
Box 3 Other investigations recommended in the assessment of pulmonary hypertension
Respiratory:
6 min walk test
arterial blood gases in room air
lung function (including FEV1, FVC, TLC, FRC, RV, TLCO, DLCO, KCO)
nocturnal oxygen saturation monitoring
Cardiology:
ECG
echocardiogram
cardiac catheterisation (including right heart catheterisation with saturations and haemodynamics, and acute pulmonary vasoreactivity study as appropriate)
Blood investigations include:
routine biochemistry and haematology
thrombophilia screen in CTEPH
thyroid function
autoimmune screen (including anti-centromere antibody, anti-SCL70 and U1 RNP, phospholipid antibodies)
hepatitis serology
serum angiotensin converting enzyme
HIV
Urine:
β-HCG (women)
5.5.1. Cardiac and lung imaging
The purpose of cardiac imaging is to determine if a cardiac cause of pulmonary hypertension is present, and assess severity. The particular advantage of non-invasive imaging is that it is safe, quick and simple to follow serially. Echocardiography can be performed at the bedside. Transoesophageal echocardiography is not routinely required but may be needed if congenital heart disease is suspected.
Lung imaging is used to detect CTEPH or parenchymal lung disease. Different techniques are used to achieve a diagnosis of CTEPH52 (fig 3). Parenchymal lung disease can be assessed on high resolution CT (table 3).

RECOMMENDATIONS
19. New patients require detailed investigation including cardiac and lung imaging to determine the aetiology and severity of PH.
5.5.2 Exercise testing
A number of exercise test protocols have been used to assess exercise capacity although none are ideal.
The role of the 6 min walking test (6MWT) in the assessment of PH is firmly established. Guidelines describe how to perform this.53 Baseline values for distance walked correlate with functional class, pulmonary haemodynamics, cardiopulmonary exercise testing (CPET) variables and survival.54 Serial values have proven to be a useful outcome measure in the majority of drug trials in PH.2 It is not an absolute change in walk distance following treatment that is predictive of survival but achieving a threshold distance.15 The sensitivity to change diminishes as the distance walked increases, particularly >450 m, and consequently may be less useful for patients in WHO functional classes I and II.55
Variants of the 6MWT include the shuttle test and endurance shuttle.56 ,57
The incremental CPET is well standardised although technically more complex to perform.58 Baseline values have been shown to be predictive of disease severity and survival (including peak oxygen consumption, systolic blood pressure at peak exercise, the ventilatory equivalent for carbon dioxide and end-tidal carbon dioxide partial pressure).59 ,60 The test can help with differential diagnosis because patients with PH show characteristic changes.59 ,61 The significant disadvantage with the test is that it has not been proven useful as a serial measurement in drug trials.62–64
RECOMMENDATIONS
20. The 6MWT is the preferred exercise outcome measure for use in assessing patients with PH both at baseline and subsequent visits and should be performed according to American Thoracic Society (ATS) guidelines.
21. A baseline CPET may be useful in selected cases to confirm that exercise limitation is due to PH and to delineate further disease severity and prognosis.
5.5.3 Lung function
Comprehensive dynamic and static lung function testing is able to detect the presence of coincident obstructive or restrictive lung disease. The classical picture in PAH without coexistent lung disease is normal spirometry and lung volumes, sometimes with mild restriction, but decreased diffusing capacity. Normal lung function does not preclude PH.
RECOMMENDATIONS
22. Lung function testing should include the measurement of spirometry, static and dynamic lung volumes and DLCO.
5.5.4 Biomarkers
Several biomarkers have been shown to be useful markers of heart disease. Brain natriuretic peptide (BNP) is released from ventricular myocytes in response to increased wall tension. It is recognised as a predictor of mortality, disease progression and response to therapy in PAH and CTEPH.65–67 ProBNP is the prohormone which is cleaved into active BNP and more stable N-terminal fragment, NT-proBNP. The levels of BNP and proBNP are dependent on age, sex, glomerular filtration rate and obesity.66 ,68–70
BNP and/or proBNP have been shown to be elevated in IPAH, PAH associated with scleroderma, systemic-to-pulmonary shunts, and PH with interstitial lung disease, chronic obstructive pulmonary disease (COPD), and CTEPH.66 ,67 ,71–75 Baseline and/or serial changes in BNP and NT-proBNP correlate with survival and surrogate markers such as pulmonary haemodynamics, functional class and 6MWT distance.65 ,66 ,76–78 Data on troponin are too limited to make any recommendations at present.79
RECOMMENDATIONS
23. Baseline plasma NT-proBNP is a useful prognostic marker in PAH patients without significant renal or left ventricular impairment.
5.5.5 Right heart catheterisation
Right heart catheterisation should be undertaken in new patients after other investigation results have been reviewed in order to determine exactly which measurements are needed (box 4). It is also used to answer specific clinical questions during follow-up. Cardiac output should be measured either by thermodilution or the Fick method, the latter only when oxygen consumption is measured. Exercise haemodynamics may be helpful to evaluate borderline PH and left heart disease.
Box 4 Measurements typically made at right heart catheterisation
Pressure measurements should be made in the following places:
systemic artery
pulmonary capillary wedge (or left ventricular end-diastolic pressure if not obtainable)
pulmonary artery
right ventricle
right atrium
(left atrium if entered via a patent foramen ovale or atrial septal defect)
Blood samples for oximetry should be taken from:
systemic artery
(left atrium if entered)
pulmonary artery (take 3 saturations and average results)
Derived variables to be calculated:
cardiac output and index
pulmonary and systemic vascular resistances
A vasoreactivity study is undertaken to determine suitability for high dose calcium antagonist therapy in those groups of patients who are known to benefit from such therapy. These studies are performed using inhaled NO (the agent of choice), or an infusion of intravenous epoprostenol or adenosine.2 A positive response is apparent when the mean PAP falls at least 10 mm Hg to <40 mm Hg with either an increase or no change in cardiac output. A vasoreactivity study is contraindicated in PAH associated with significant venous or capillary involvement because of the risk of pulmonary oedema.
RECOMMENDATIONS
24. Right heart catheterisation is essential during the initial investigation of new patients.
25. A vasoreactivity study should be performed in patients with IPAH, FPAH, CTD APAH (excluding SSc APAH), and anorexogen-induced APAH. Only responders should be treated with high dose calcium channel blockers.
6. OBJECTIVES AND PRIORITIES FOR TREATMENT
6.1 Anticoagulation
Vascular thrombotic lesions have been identified with high prevalence at post-mortem in patients with IPAH80 ,81 and other forms of PAH.82 Although a relationship between thrombotic lesions, age and disease duration83 have been suggested this is not a universal finding.17 Thrombosis appears uncommon in children. Abnormalities in coagulation and fibrinolytic pathways, and platelet function have also been demonstrated.84–86 Clinical studies are limited. Two retrospective81 ,87 and one small non-randomised prospective study88 have associated anticoagulation use with a survival benefit. These studies have been conducted almost exclusively in patients with IPAH. Randomised controlled trials are needed in patients with PAH associated with other diseases where the risk benefit ratio of anticoagulation is not known.
RECOMMENDATIONS
26. Anticoagulation with warfarin is recommended in patients with IPAH and CTEPH in the absence of contraindications. The international normalised ratio (INR) should be maintained between 2 and 3. For IPAH patients with a higher than normal bleeding risk an INR of 1.5 to 2.5 is suggested.
27. Anticoagulant therapy is recommended in Ssc APAH although this recommendation is purely consensus based.
6.2 Oxygen therapy
Oxygen administered acutely has been demonstrated to reduce PVR in both hypoxic and non-hypoxic patients with PH. There are no randomised data available to suggest that long term oxygen therapy (LTOT) is beneficial in PAH. There are data showing that nocturnal oxygen therapy does not modify the natural history of advanced Eisenmenger syndrome89 (see section 6.4.8.3). This does not exclude the possibility of benefit from LTOT in other patient groups with PH since it is an increase in alveolar oxygen which leads to a reduction in pulmonary vascular resistance. Arterial oxygenation is not a good reflection of alveolar oxygenation in Eisenmenger physiology.
Based on the limited evidence from studies with COPD,90 ,91 oxygen should be prescribed in accordance with the BTS Working Group on Home Oxygen Services.92 When arterial oxygen pressure (Pao2) is consistently at or <8 kPa (breathing room air) during a period of clinical stability, oxygen should or may be prescribed for at least 15 h a day (including night time) to achieve a Pao2 of >8 kPa. Where daytime oxygenation is satisfactory, nocturnal oxygenation should be assessed and oxygen prescribed if mean overnight saturations are <90% to achieve a mean saturation greater than this. There can be no recommendation for oxygen in PAH associated with congenital heart disease.
Arterial hypoxaemia can contribute to breathlessness on exertion through stimulation of the peripheral chemoreflex. The prescription of ambulatory oxygen should follow the recommendations of the BTS document.92 Consequently, the patient will qualify for ambulatory oxygen if there is evidence of symptomatic benefit and correctible desaturation of >4% to <90% on a 6MWT.
There have been no studies using flight simulation to determine which patients require oxygen during air travel, but given the known physiological effects of hypoxia, it seems currently prudent to consider in-flight oxygen for all patients with significant pulmonary hypertension. A flow rate of 2 l/min will raise inspired Po2 to sea level values.
RECOMMENDATIONS
Except with congenital heart disease:
28. All patients should have nocturnal oxygen saturation monitoring at initial assessment and thereafter when clinically indicated.
29. Oxygen should be administered to maintain daytime and nocturnal Pao2 >8 kPa.
30. Ambulatory oxygen can be considered in those with correctable exercise desaturation of >4% to <90% for symptomatic benefit.
31. In-flight supplemental oxygen should be considered for all patients in WHO functional class III and IV or those with resting oxygen saturations <95%.
6.3 Supportive medical therapy
Assisting patients to adapt to the uncertainty associated with chronic, life shortening disease is essential if they are to adjust successfully to the demands of their illness and its treatment. The health care team needs to be highly skilled in managing the burden and impact of this disease, and its complex and intrusive therapies at both physical and psychological levels.
Patients with PH often feel isolated by their diagnosis93 and many seek help from support groups for numerous reasons including learning about their illness, sharing coping strategies with others who have similar health problems, sharing their experiences, and gaining emotional support. Encouraging patients and their family members to be part of support groups can have positive effects on coping, confidence, outlook and relationships.94 Nationally the UK pulmonary hypertension patient group (The Pulmonary Hypertension Association UK, http://www.pha-uk.com) offers support to patients and their carers. Patients may also access the NHS Expert Patients Programme for people living with long term chronic ill health through their local GP surgery or library, and their local pulmonary hypertension support group where this exists.
RECOMMENDATIONS
32. Patients with PH should be managed by an experienced multiprofessional team with the required skill and expertise to meet the holistic needs of the patient and their carers. These needs include information about the disease and its prognosis, education and support in managing complex drug therapies, psychological, social and spiritual support, and access to local support in the community.
33. Patients should be encouraged to join a support group.
6.3.1 Family planning
Pregnancy is associated with a high risk of maternal death. The WHO identifies PH as a contraindication to pregnancy and advises discussing termination in the event of pregnancy.
For contraception progesterone only preparations such as medroxyprogesterone acetate and etonogestrel are highly effective.95 The Mirena coil is also effective but 5% of women may have a vasovagal reaction96 which can have potentially fatal effects in the setting of reduced cardiovascular reserve. Sterilisation is rarely used in this population due to the operative risks and higher failure rate. Bosentan is an enzyme inducer and may reduce the efficacy of hormonal contraceptive preparations (see section 6.4.7).
Some patients who become pregnant choose to continue their pregnancies even when they are informed of the high risk. Despite a variety of approaches to its management97–99 the mortality remains high. Early treatment with disease-targeted therapy100 ,101 may improve the chances of maternal survival. With timely admission to hospital, planned elective delivery102 and incremental regional anaesthesia with close cooperation of a PH multidisciplinary team, a successful outcome for mother and fetus may be possible although maternal mortality remains high.103
RECOMMENDATION
34. Patients with PH should be counselled regarding the very high risk of pregnancy (>30% mortality) with clear contraceptive advice. They should be offered early termination of pregnancy if the pregnancy is unwanted.
35. If a patient becomes pregnant termination of pregnancy should be discussed. When patients are fully informed and understand the risks of proceeding with pregnancy, treatment with disease-targeted therapies for PAH represents a realistic option and may improve the chances of maternal survival.
6.3.2 Physical activity
Advice regarding physical activity is empirical in PH and based on consensus opinion. A recent study has demonstrated an improvement in exercise capacity in patients who took part in a training programme.104
RECOMMENDATIONS
36. Patients should be encouraged to be as active as their symptoms allow. Mild breathlessness is acceptable but patients should be advised to stop exercising if they become moderately or severely breathlessness, or develop exertional dizziness or chest pain.
6.3.3 Heart failure and arrhythmias
Heart failure gives rise to fluid retention which is improved by diuretics, although no randomised controlled trials exist on the use of diuretics in PH.
Digoxin has been shown to improve cardiac output acutely in IPAH,105 although its efficacy is unknown when administered chronically.
Atrial flutter and other tachyarrhythmias are often poorly tolerated and may present with worsening heart failure or syncope. Treatment with an appropriate therapy after verification of the arrhythmia is recommended. Note that β-blockers are poorly tolerated in PH.106
RECOMMENDATIONS
37. Diuretics are indicated to reduce fluid retention.
38. Digoxin may be beneficial in heart failure and should be considered in patients in sinus rhythm who remain symptomatic on medical therapy.
39. Acute arrhythmias require prompt management with the aim of restoring sinus rhythm and preventing recurrence of the arrhythmia.
6.3.4 Immunisations
Patients with PH are prone to infections which are often poorly tolerated because of their reduced cardiovascular reserve.
RECOMMENDATION
40. Patients should be offered immunisation against pneumococcal pneumonia and annual immunisation against influenza.
6.4 Disease-targeted therapies for PAH
The aim of therapy is to improve survival, symptoms and QoL. On the basis of a series of randomised trials, epoprostenol, iloprost, treprostinil, bosentan, sitaxsentan and sildenafil are available as monotherapy in some forms of PAH. These drugs offer not only improved symptom control, exercise capacity, QoL and haemodynamics, but also the prospect of extended survival. Survival is closely related to WHO functional class and exercise capacity.15 ,107
6.4.1 Therapeutic classes of drugs
Only therapies that are used in current clinical practice in the UK and Ireland have been considered. Since many reviews of drug trials in PAH have been published, we have chosen to tabulate those studies published before November 2006. For brevity these trials are not discussed further in the text. Tables 4–⇓⇓⇓⇓⇓⇓11 include all randomised trials and those non-randomised trials considered by the consensus meeting to have significant impact based on the number of patients in the trial, study design and data collection. They have not been selected on the basis of a positive or negative result.
Entry criteria into the randomised trials were similar across studies: 6MWT distance >100–150 m and <450–500 m (except STRIDE-1 study which had no exercise limitations), mean PAP >25 mm Hg, PCWP ⩽15 mm Hg and PVR >240 dynes/s/cm5.
There are no large comparative studies between the drugs and therefore our recommendations are based upon a consensus of UK and Irish specialists and the 2004 European Society of Cardiology guidelines.2
Survival has been examined up to 5 years in open label studies. In these tables, data are only reported up to 2–3 years owing to the small number of patients beyond these time points. Some survival studies compare observed survival against NIH predicted survival.14 It is not clear whether this formula can accurately predict the current natural history of IPAH and its results should be interpreted with caution.
6.4.2 Calcium channel blockers
High dose calcium channel blockers are beneficial in a subset (<10%) of patients with IPAH, FPAH and anorexigen APAH who demonstrate a positive vasoreactivity response at right heart catheterisation and have improved 5 year survival.88 ,108–110 In these patients treatment is started with slowly titrated high dose calcium channel blockers (for example, up to 240 mg/day of nifedipine or up to 900 mg/day of diltiazem). If there is no improvement after 1 month or patients are unable to achieve WHO class I or II with associated improvement in haemodynamics over 3 months, then patients should be treated as non-responders. Only 54% of those who respond acutely will maintain a sustained response to calcium channel blockers.110 Patients who have not undergone a vasoreactivity study or those with a negative study should not be started on these agents.111
RECOMMENDATIONS
41. Patients with IPAH, FPAH and anorexigen APAH and a positive vasoreactivity response should be treated with high doses of calcium channel blockers. Since only half will respond chronically, it is important to ascertain normalisation or near-normalisation of PAP at follow-up.
42. If patients taking high dose calcium channel blockers demonstrate no clinical improvement after 1 month or are unable to achieve functional class I or II with associated improvement in haemodynamics over 3 months, then they should be treated as non-responders.
6.4.3 Prostanoids
Prostanoids are analogues of prostacyclin and include epoprostenol, iloprost and treprostinil. They must be given by infusion or aerosolised for inhalation. Dosing requires balancing symptom control with side effects (including headache, flushing, nausea, vomiting, loose bowel motions, jaw pain and limb pain) and needs to be managed on an individual basis. There is no prescribed upper limit to dose. In most patients the dose requires progressive uptitration over time. There is a risk of local and systemic sepsis in patients on infusions. Patients must be able to manage an infusion or frequent inhaled treatment at home.
It is not possible to draw firm conclusions about the relative efficacy of different prostanoids. The choice of therapy is determined by patient and physician choice which in turn is affected by differences in the half-life of the drugs, side effect profile, complexity of the delivery system, patient acceptability and the patient’s home circumstances. In some patients non-licensed therapies are preferred for these reasons.
The results of randomised trials11 ,110 ,112–116 are shown in table 4 and non-randomised trials15 ,107 ,117–126 in table 5. Their use is described in fig 4.
6.4.4.Phosphodiesterase 5 inhibitors
PDE 5 inhibitors are the newest class of disease-targeted therapy in PAH and there is less long term experience than prostanoids or endothelin receptor antagonists (ERAs). Only sildenafil is available for use in PAH. While sildenafil has been licensed for use at 20 mg three times daily, longer term survival data has been collected at 80 mg three times daily.
The results of randomised trials127 ,128 are shown in table 6 and non-randomised trials129 ,130 in table 7.
6.4.5 Endothelin antagonists
Bosentan, an ETA and ETB receptor blocker, and sitaxsentan, a selective ETA receptor blocker, are available in the UK.
Patients taking ERAs require monthly liver function tests to monitor transaminases which may rise and progress to liver failure unless the dose is reduced or the drug discontinued.
The results of randomised trials63 ,131–135 are shown in table 8 and non-randomised trials136–143 in table 9.
6.4.6 Combination therapy
The results of randomised trials144–146 are shown in table 10 and non-randomised trials147–150 in table 11.
The optimal management for those patients who exhibit clinical deterioration despite targeted monotherapy remains a matter of debate.9 In the event of worsening functional status and haemodynamics, the approach of combining different agents to augment the clinical response has a strong rationale and has already been widely adopted by clinicians in most centres in Europe and the USA.150
6.4.6.1 Rationale for combination therapy
The use of combinations of drugs acting on distinctly different pathways involved in the disease in order to maximise clinical gain for patients with pulmonary hypertension is an emerging concept3 ,151 which has gained acceptance in published guidelines.2 ,6 The rationale can be summarised:
1. Multiple pathways involved
The notion that a unifying molecular mechanism might be critical to the development of PAH seems improbable given that there are numerous distinct clinical syndromes, environmental factors and genetic abnormalities which predispose to the disorder.152 Furthermore, a number of defects have been demonstrated in distinct pathways that have proved amenable to targeted therapies in clinical trials (that is, NO, prostacyclin and endothelin pathways). Consequently, it is unlikely that employing treatments that act on a single pathway will be consistently successful. A broad based approach targeting multiple pathways is more likely to be effective.153
2. The precedent for combination therapy in medicine
Combination therapy is often necessary in order to optimally control systemic hypertension, while in a variety of other diseases such as cardiac failure, cancer and HIV infection, combination therapy has become standard care supported by the highest levels of evidence. The potential for targeting multiple pathways at the time of initial diagnosis for patients with advanced disease makes intuitive sense. A treatment algorithm that includes combinations of individually successful drugs has merit even in the absence as yet of robust support from the medical literature.
3. The lack of a durable response to monotherapy
Many patients will have a suboptimal response or develop tolerance to an initial therapeutic regimen.15 ,107 ,114 ,116 ,127 ,131 Follow-up beyond the first 3 months of oral therapy shows a proportion of patients deteriorate. A well-recognised drawback of long term epoprostenol therapy is tachyphylaxis154 and repeated dose escalation is frequently hindered by the onset of disabling side effects at high doses. The lack of durable response does not indicate that monotherapy is failing to have a clinically important effect in slowing disease progression. Indeed withdrawal of the original disease-targeted therapy after addition of a second therapy may result in acute clinical deterioration even in stable patients.155 There are no formal trials of, or tested protocols for, withdrawing the original therapy. Reluctance of clinicians to withdraw the original therapy comes from the precarious clinical state of patients who are failing monotherapy as well as evidence that prostacyclin withdrawal results in clinical deterioration which cannot be rescued by restarting therapy.
4. Synergies between agents
By exploiting molecular interrelationships between individual therapeutic targets, it may be possible to improve overall treatment efficacy and minimise risk of toxicity by using reduced dosages of individual agents. PDE inhibitors are responsible for limiting the adenylate cyclase regulated actions of epoprostenol.156 The addition of PDE inhibitors may augment and/or prolong the vasodilatory effects of prostanoids through “crosstalk” between the cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) pathways. The addition of sildenafil in patients refractory to epoprostenol is associated with enhanced levels of cAMP, an effect mediated via inhibition of PDE-3 by cGMP.157 This effect in turn causes upregulation of NO production and further increases in cGMP concentrations. Sildenafil, when added to aerosolised iloprost, causes a greater reduction in PVR than either drug alone.147 ,158 Co-treatment with sildenafil and beraprost may also result in greater increases in plasma concentrations of cAMP and cGMP than with either drug alone.159 Bosentan and sildenafil have synergistic effects on haemodynamics and mortality in animal models.160 Since endothelin suppresses NO production,110 ,161 ERAs upregulate NO production and synergise sildenafil.147 ,162
6.4.6.2 Clinical evidence for use of combination therapy
To date there have been relatively few prospective trials conducted to appraise the merits of combining drugs with different modes of action for the treatment of PAH. Many authors have reported successful outcomes with this approach in animal models of PH as well as in observational series and non-randomised studies. Most of the studies currently available are of a retrospective or observational nature, describing experiences with small numbers of heterogeneous patients (often from a single centre) without matched controls characterised. Investigators have mostly chosen to investigate IPAH, CTD APAH or anorexigen induced APAH, limiting the applicability of results to other categories of PH. A number of well-designed studies are now underway (Appendix 1) that should help clarify the potential benefits of a variety of different combination strategies in the next 1–3 years.
A comprehensive listing of published studies is included in this manuscript (tables 4–⇑⇑⇑⇑⇑⇑11). There are only four possible combinations of currently available agents: ERAs and prostanoids, ERAs and PDE 5 inhibitors, PDE 5 inhibitors and prostanoids, or all three drug types together. Because there are four types of prostanoids and two ERAs the combinations can become complex within these four groups.
6.4.6.3 Approaches to combination therapy
Given the limited amount of conclusive data pertaining to combination treatment, published guidelines have so far made no specific recommendation to guide clinicians on the next step to improve the status of their patients who fail monotherapy.
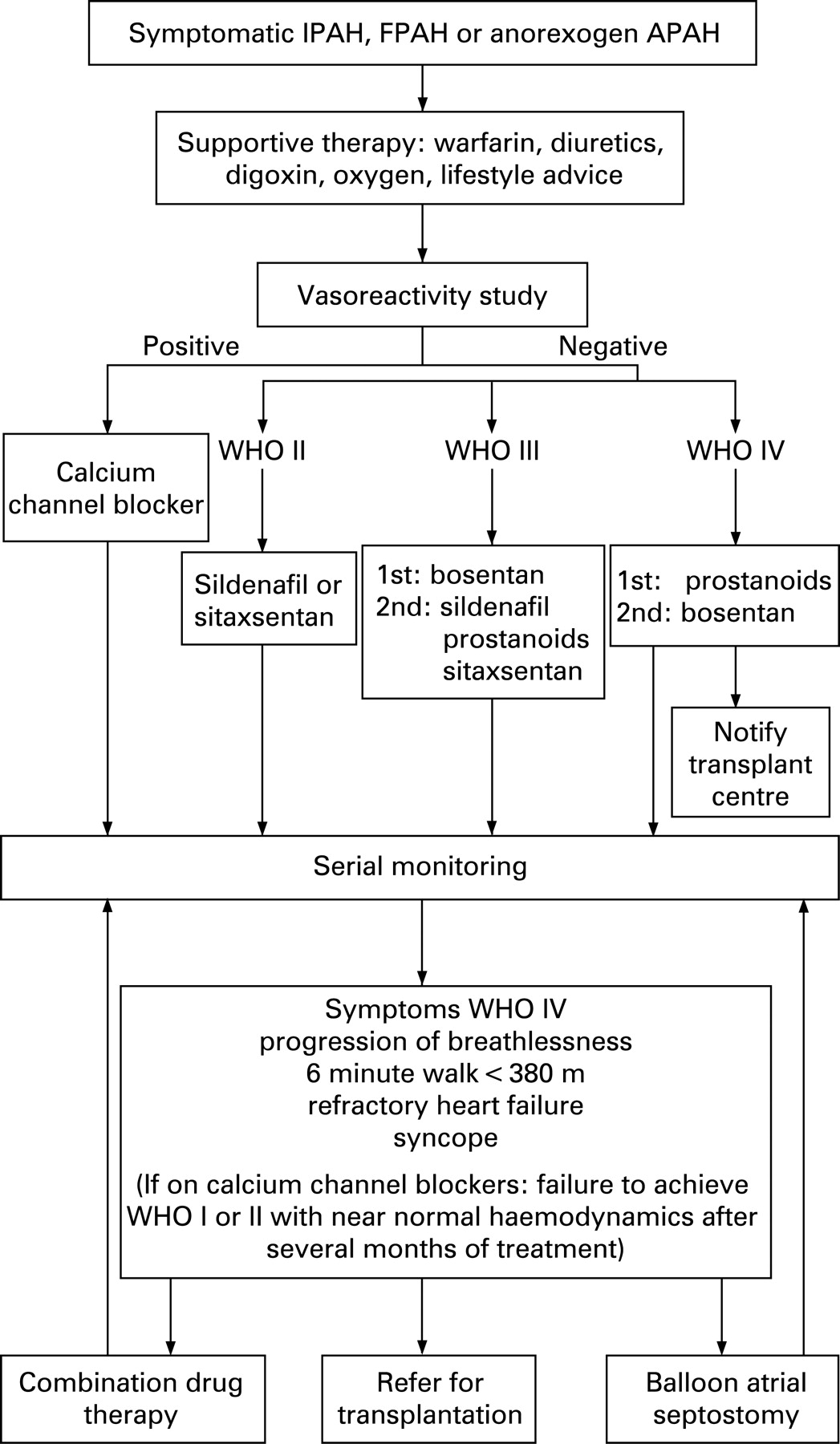
The most commonly applied combination therapy approach to date has involved the addition of a second drug where patients deteriorate (or fail to sufficiently improve) despite optimal doses of an initial therapy. The algorithms in figs 4 and 5 indicate commonly used clinical criteria for considering combination therapy. These criteria were chosen because they indicate a particularly poor prognosis and can be documented.

For patients who present in WHO functional class III the most common combination is the addition of a second oral agent (sildenafil to an ERA or an ERA to sildenafil). In the UK this accounts for 65% of combination therapy prescriptions. In small studies this approach has been shown to be effective in improving functional class, 6MWT distance and peak oxygen consumption.149 ,163 ,164
The addition of a parenteral prostanoid to the original oral therapy increases the complexity of administering treatment. It is indicated in the event of rapid deterioration, especially when expected survival time is limited. The combination of a prostanoid plus sildenafil accounts for 23% of combination therapy prescriptions in the UK and confers added clinical benefits to monotherapy compared with either class of drugs alone.147 ,148 ,158 ,165
Although evidence suggests that the combination of bosentan and epoprostenol may have limited benefit,145 the addition of bosentan to treprostinil improved functional class, exercise capacity and haemodynamics,148 and the addition of inhaled iloprost to bosentan monotherapy improved exercise capacity.146 Prescription of a prostanoid plus an ERA accounts for 12% of combination therapy prescriptions in the UK.
Given the likelihood of eventual monotherapy failure, another approach is to commence two (or more) treatments simultaneously at the time of diagnosis. This approach is occasionally applied in patients in WHO functional class IV where survival without therapy is measured in months as evidenced by exercise capacity and haemodynamics.
While awaiting the results of further clinical trials a consistent approach to combination therapy is urgently required in view of the risk of fatal outcome. The trigger to consider combination therapy should be part of a goal-oriented strategy to assist timing of treatment escalation.150 Individuals who demonstrate an inadequate exercise tolerance despite monotherapy for 3–4 months should be considered for additional therapy as outlined in figs 4 and 5. All patients should be offered a second agent as part of a clinical trial where possible. There are a number of trials enrolling patients with PAH who are on monotherapy with either bosentan or sildenafil (Appendix 1).
RECOMMENDATIONS
43. Individuals who demonstrate an inadequate response to monotherapy (see figs 4 and 5) should be considered for a combination of two or more disease-targeted therapies.
44. Where combination therapy is to be used, patients should be entered into a clinical trial where possible.
45. In the absence of a clinical trial, there is sufficient expert consensus to proceed with combination therapy while the patient’s response to treatment is carefully monitored with consistent measures linked to national audit.
6.4.7 Drug interactions of disease-targeted therapies for PAH
There is the potential for clinically significant drug interactions from combining some therapies (table 12). In addition to pharmacokinetic interactions that alter drug concentrations and may affect efficacy and increase toxicity, pharmacodynamic interactions in the systemic vasculature may lead to hypotension and systemic side effects such as dizziness.
Bosentan is an inducer of CYP3A4, a hepatic enzyme involved in the metabolism of many drugs, as well as being a substrate for this enzyme. Co-administration of bosentan and sildenafil (a CYP3A4 substrate) in patients with PAH has been shown to reduce plasma sildenafil levels by 50%.166 Conversely, bosentan levels are increased by about 50%, presumably by competition with sildenafil for metabolism. The net effect of these changes on response of patients to the combination has not been determined but the implications are: (1) the possibility of hepatic toxicity from higher plasma bosentan levels; and (2) lack of effect from low dose sildenafil (that is, 20 mg dose regimen) when given with bosentan. Bosentan has been reported to reduce the level, and so efficacy, of other CYP3A4 substrates, such as simvastatin, oral oestrogens and ciclosporin. In contrast, CYP3A4 inhibitors, such as ketoconazole and ciclosporin, increase bosentan levels; co-administration of bosentan and ciclosporine is contraindicated. Bosentan induces CYP2C9 and increases warfarin metabolism.
Sitaxsentan is an inhibitor of this enzyme and co-prescription with warfarin requires a reduction in warfarin dose to maintain a therapeutic INR. Sitaxentan is a weak inhibitor of CYP3A4/5. It produces an increase in oestrogen levels when given with oral oestrogen contraceptives, and a small but clinically insignificant elevation of sildenafil when given with this drug.
Sitaxentan levels are increased sixfold when given with ciclosporin and co-administration of these drugs is contraindicated.
Sildenafil, as discussed above, is a CYP3A4 substrate and levels are increased by inhibitors of this enzyme, such as erythromycin, ketoconazole and several HIV protease inhibitors, such as ritonavir and saquinovir.
6.4.8 Recommendations and strategies for the use of disease-targeted therapies
6.4.8.1 Idiopathic, familial and anorexogen-induced PAH
While patients in WHO functional classes III and IV were included in early trials, recent trials have seen the inclusion of functional class II patients. There is no evidence to support the treatment of functional class I and this has been excluded from the algorithm (fig 4).
The benefit of identifying responders to calcium channel blockers has been covered in section 6.4.2. Most patients in WHO functional class III are commenced on oral therapy as first line treatment as distinct from functional class IV where evidence for prostanoids predominates. In some severely ill patients in functional class IV, most pulmonary hypertension specialists are unwilling to undertake an acute vasoreactivity study and would institute an intravenous epoprostenol infusion immediately.
RECOMMENDATIONS
46. Patients with IPAH, FPAH or anorexigen-induced PAH should be managed according to the algorithm in fig 4.
6.4.8.2 Connective tissue disease
Data on the treatment of CTD APAH have been acquired from subpopulations of larger studies and are more limited than IPAH. An algorithm for CTD APAH management is shown in fig 5.
6.4.8.2.1 Supportive therapy
Oxygen therapy may alleviate moderate hypoxia. The use of diuretics and digoxin may benefit some patients.167
6.4.8.2.2 Calcium channel blockers
The prevalence of a vasodilator response in CTD APAH is reported between 2–5%.168 ,169 While reductions in PVR are common these do not appear to predict outcome in SSc.42 None of these studies has reported a clear correlation between response to calcium channel blocker therapy and vasodilator response in the setting of CTD. Tolerance of high dose calcium channel blockers is unusual in SSc APAH.42
RECOMMENDATION
47. Routine vasodilator testing in patients with SSc APAH is not mandatory since this does not identify a population who will benefit from calcium channel blockers.
6.4.8.2.3 Prostanoids
Epoprostenol improves 6MWT distance in patients with severe SSc APAH.112 There are no long term data showing improved prognosis. There was no improvement in outcome for SSc APAH, half of whom were treated with epoprostenol,170 when compared to an untreated population171 10 years earlier.
Not all prostanoids appear to be effective in SSc APAH.62 ,114 ,172 Although this population were less tolerant of higher doses of treprostinil (mean 8.4 ng/kg/min after 12 weeks), they exhibited haemodynamic benefit (increase of cardiac index by 0.2 l/min/m2 and reduction of indexed PVR by 320 dynes/s/cm5). There was a trend toward 6MWT distance improvement.173 Inhaled iloprost showed little benefit in the CTD APAH subpopulation.114 More data on SSc APAH are required to determine the future roles of these agents.
6.4.8.2.4 Endothelin receptor antagonists
Endothelin levels are elevated in the dermis and internal organs of SSc patients174 ,175 as well as lung tissue.176 The fundamental role of endothelin in fibrotic, mitogenic and proliferative activity, and vasoconstriction suggests that ERAs may influence the pathobiological processes underlying CTD APAH.177
In the SSc subgroup of double-blind, placebo-controlled trials, bosentan prevented a decline in functional ability seen with placebo patients.133 Bosentan improved 6MWT distance, delayed the time to clinical worsening and reduced dyspnoea. The 1 year survival at 81% was the best reported survival data at the time. In a single centre historical control registry, bosentan therapy was associated with a marked improvement in survival when compared to previously available therapy.141 The safety of this therapy has now been demonstrated in the TRAX registry which includes nearly 1500 patients with CTD APAH with an average follow up of 9 months.178
Post hoc analysis of sitaxsentan trials found 119 patients with CTD (63 SSc, 22 MCTD, 25 systemic lupus erythematosus (SLE)) APAH, 58 of whom received placebo, 61 were treated with sitaxsentan 100 mg once daily, and small numbers who received either 50 mg or 300 mg.179 For those taking the 100 mg dose, the net improvement in 6MWT distance was 38 m, similar to the improvement seen in IPAH (p = 0.042).
6.4.8.2.5 Phosphodiesterase inhibition
The SUPER-1 trial127 shows a similar magnitude of benefit with sildenafil to IPAH in the subgroup with CTD APAH.180 In this trial 84 patients had CTD APAH (including 38 SSc, 12 CREST (calcinosis, Raynaud’s phenomenon, oesophageal dysmotility, sclerodactyly, and telangiectasia), 19 SLE and 8 MCTD), of whom 32 were in WHO functional class II, 51 class III and 1 class IV. The 6MWT distance decreased by 13 m in the placebo group, but increased by 42 m, 36 m and 15 m in the 20, 40 and 80 mg three times daily groups, respectively. There are no long term follow data for the CTD subgroup.
RECOMMENDATION
48. ERAs should be first line therapy for patients with CTD APAH until long term data for other treatment modalities show that they have a comparable effect on survival.
6.4.8.2.6 Combination therapy
There are no data on combination therapy in the CTD PAH population. Only 10% of these patients achieve WHO functional class II, mean PAP <35 mm Hg and NT-proBNP <400 pmol/ml.78 Most have some response, but with a first year mortality of 20%, and 70% remaining in functional class III or IV, the natural history of the condition has not been sufficiently altered to deliver a satisfactory outcome. Current consensus-based clinical practice is illustrated in fig 5 and when combination therapy is used, sildenafil is added to bosentan or sitaxsentan. The next step is to either add inhaled iloprost or to switch to intravenous prostanoids.
6.4.8.2.7 Transplantation
Transplantation should be considered but is frequently not offered to patients with systemic conditions. While the results of transplantation in a very carefully selected group of patients with connective tissue disease is the same as for the population with interstitial lung disease alone, many potential candidates may not be suitable because of their associated comorbidities such as severe oesophageal dysfunction.
RECOMMENDATION
49. Video-fluoroscopic assessment of swallowing should be undertaken as part of the assessment of transplantation in SSc APAH.
6.4.8.3 Adults with classical Eisenmenger syndrome
Eisenmenger syndrome is defined as severe PAH associated with a large and non-restrictive intra- or extra-cardiac shunt which with time leads to reversal of flow and cyanosis. Although annual mortality rates for Eisenmenger patients are relatively low compared to other forms of PAH, median survival is reduced by at least 20 years and is worse in patients with complex cardiac anatomy.38 ,181 ,182 A more favourable clinical course than IPAH can be anticipated in Eisenmenger patients due to (a) a naturally occurring right-to-left shunt (sustaining systemic cardiac output, albeit at the expense of cyanosis) and (b) right ventricular remodelling occurring over a long time. Despite the varied and often complex underlying cardiac anatomy and physiology, pulmonary vascular histological changes are remarkably similar to other forms of PAH, and most patients are symptomatic primarily with breathlessness.183 ,184
Secondary erythrocytosis is universal in patients with Eisenmenger syndrome. Haemoglobin concentration is inversely related to oxygen saturation only in iron-replete patients.182 Furthermore, iron deficiency closely correlates with venesection, often performed for “symptoms of hyperviscosity” or a haematocrit >65%, although many of these symptoms are also common in patients with iron deficiency. Patients with lower haematocrit have a lower exercise capacity and phlebotomy itself is associated with a higher and not lower incidence of cerebrovascular accidents.185 It is not justified to recommend routine phlebotomy in these patients. Iron deficiency should be promptly identified and corrected and an “upper limit” of haematocrit or haemoglobin should not be set.185
Many patients are anticoagulated with warfarin or given antiplatelet therapy to treat or prevent intravascular thrombosis which is common in these patients.186 Similarly, some patients are treated with LTOT at night on an individual basis, despite conflicting evidence as to a clinical benefit.89 NO, prostanoids and transplantation have been shown to be effective in improving functional class, but they are invasive and associated with problems of optimal timing and patient selection.125 ,187 ,188
Bosentan has been studied in a randomised double blind placebo controlled study (BREATHE-5) and significantly improved haemodynamics and exercise capacity without adversely affecting systemic arterial oxygen saturation in WHO functional class III patients.134 Improvements in exercise capacity were maintained mid to long term in an open-label extension study.189 Figure 6 shows an algorithm for the management of patients with Eisenmenger syndrome.

RECOMMENDATIONS
In grown-up congenital heart disease:
50. Patients with Eisenmenger syndrome have a multi-organ disease and should be managed at least in part in a grown-up congenital heart disease (GUCH) centre.
51. Each GUCH centre should have formal links to a designated pulmonary hypertension centre.
52. Iron deficiency should be corrected. Venesection should not be used routinely.
53. No specific recommendations can be given for general use of anticoagulation, antiplatelet or oxygen therapy, although all of them may be used on an ad hoc basis. INR monitoring requires specialist expertise and where anticoagulation is used haematology advice should be sought.
54. Patients in WHO functional class III should be considered for disease-targeted therapies for which there is evidence of efficacy and safety supporting the use of oral bosentan.
55. Transplantation should be considered for Eisenmenger patients remaining in WHO functional class IV.
6.4.8.4 Veno-occlusive disease and pulmonary capillary haemangiomatosis
The relationship between IPAH and pulmonary veno-occlusive disease (PVOD) and pulmonary capillary haemangiomatosis (PCH) is unclear. Given reports of familial occurrences in all three conditions190–192 and the identification of a BMPR2 receptor gene mutation in a patient with PVOD,193 it has been suggested that PVOD and PCH may represent variants of IPAH in which the primary lesion affects the venous and capillary regions of the pulmonary vascular bed.2
The presentation of PVOD or PCH is often identical to IPAH, although some clinical features can be used to distinguish between them including more pronounced respiratory failure, digital clubbing and bibasal crackles.194 High resolution CT scan of the thorax is the most discriminatory non-invasive test. The presence of centrilobular ground glass opacities, septal lines and mediastinal lymphadenopathy are the most predictive features for PVOD or PCH.195 It has also been suggested that bronchoalveolar lavage showing haemosiderin laden macrophages can be a useful discriminatory test as it demonstrates the occult alveolar haemorrhage that occurs in PVOD/PCH.196 Definitive diagnosis requires a surgical lung biopsy197 ,198 but this carries significant mortality.198
The use of disease-targeted therapies for PAH in PVOD and PCH is problematic. There are no controlled trials and there have been a number of case reports associating the use of vasodilator agents (principally calcium channel blockers and prostanoids) with worsened or even fatal pulmonary oedema.191 ,194 ,199–202 This is particularly true with PCH.203 Conversely, in PVOD there are case reports where the use of vasodilator agents has appeared to stabilise the condition.194 ,204–206 There are no published data on the use of ERAs in PVOD and there is only one reported case of the use of sildenafil,207 although unpublished experience with this agent has been promising. Anti-angiogenic agents such as interferon-α2a and doxycycline have been used in a small number of patients with PCH with variable reports of benefit.199 ,203 ,208–210
Most patients with PVOD die within 2 years.194 ,211 Median survival with PCH is 3 years.203 Transplantation has been successful in both PVOD and PCH,203 ,211 although a recent case report suggests PVOD may recur after transplantation.212
RECOMMENDATION
56. An acute vasoreactivity study should not be performed in patients with PVOD or PCH, although right heart catheterisation should be performed.
57. Anticoagulation may be hazardous since haemoptysis may be troublesome, particularly in PCH.
58. Treatment results with disease-targeted therapy are anecdotal. Given the poor prognosis of the condition and the reports of temporary stabilisation in some patients, disease-targeted therapies may be attempted but must be stopped if there is any indication of worsening pulmonary oedema.
59. PCH should not currently be treated with disease-targeted drugs. Anti-angiogenic agents such as doxycycline may be considered in association with laboratory measurement of angiogenic factors.
60. Consider immediate referral for transplantation at time of diagnosis.
61. Atrial septostomy is a treatment option but is often precluded by hypoxaemia.
6.4.8.5 Chronic thromboembolic disease
6.4.8.5.1 Epidemiology
CTEPH has emerged as one of the leading causes of severe PH. The incidence and prevalence of CTEPH are unknown. Originally it was thought that 0.1–0.5% of patients who survived an episode of acute pulmonary embolism develop CTEPH.213 ,214 More recent studies suggest 3.1% of patients will develop symptomatic CTEPH at 1 year and 3.8% by 2 years post-pulmonary embolism.50 The natural history of untreated CTEPH is dismal: <20% of patients survive 2 years if the mean PAP is >50 mm Hg at the time of presentation.215
6.4.8.5.2 Pathophysiology
CTEPH is characterised by intraluminal thrombus organisation and fibrous stenosis or complete obliteration of the pulmonary artery lumen.10 In the currently accepted model of CTEPH disease, acute pulmonary embolism, whether symptomatic or asymptomatic, serves as the initiating event. Following the embolism, disease progression occurs due to progressive pulmonary vascular remodelling and development of a generalised hypertensive pulmonary arteriopathy216 in those areas of the pulmonary vasculature that were spared from thromboembolic occlusion. Histopathology shows changes in the pulmonary microvasculature in CTEPH appearing similar to those seen in other forms of severe, non-thromboembolic PAH.216–218 This model explains why some CTEPH patients have severe PH out of proportion to the degree of vascular obstruction documented on pulmonary angiography. Patients with significant PH of microvascular origin but with little or no visible evidence of thromboembolic segmental vessel occlusion may not always benefit from PEA surgery.219 ,220
The mechanism underlying the development of CTEPH is not known. Two factors are important and should be distinguished: prevention of complete recanalisation of a pulmonary artery after pulmonary embolism, and the process of remodelling in small vessels. The normal pulmonary circulation carries a high fibrinolytic potential, but alteration in the fibrinolytic system has not been identified in patients with CTEPH. The only factors that have been linked to CTEPH are anticardiolipin antibodies221 and elevated factor VIII.222 Other risk factors include chronic inflammatory disease, myeloproliferative syndromes, ventriculo-atrial shunt and splenectomy.223
6.4.8.5.3 Clinical findings
Patients with CTEPH usually present with symptoms and signs of chronic PH, complaining of progressive dyspnoea on exertion and fatigue, with signs of right heart dysfunction. Others present after a single or recurrent episode of pulmonary embolism. Clinical findings are similar to other forms of PH with the addition of a bruit in the lung periphery in approximately 10% of patients. This is highly specific although of low sensitivity.
6.4.8.5.4 Investigation and general management
As the therapeutic approach to CTEPH is different to PAH, it is important to clarify the diagnosis by following the diagnostic algorithm in fig 3.224 Patients with CTEPH should receive life-long anticoagulation with warfarin in the therapeutic range of INR between 2–3.225 It is accepted worldwide practice to insert an inferior vena cava (IVC) filter in patients with CTEPH undergoing PEA surgery. There is little evidence to support this management, although there is a risk of thromboembolism in the early postoperative period until full anticoagulation is re-established. In the UK an IVC filter is inserted in most patients and there have been no adverse consequences arising from this policy to date. In females of reproductive age the IVC filter may be removed postoperatively to avoid potential problems of IVC filters in pregnancy.
6.4.8.5.5 Surgical management
The treatment of choice for symptomatic CTEPH is PEA.219 This is the only proven treatment to offer significant symptomatic and prognostic benefit.
To identify those patients who will receive the greatest benefit from surgery, the disease has been classified into four subgroups based on the operative findings. Patients with proximal disease (types 1 and 2) have a much better risk: benefit ratio from surgery226 than more distal disease. Those with a PVR >1200 dyne/s/cm5 have a higher risk of mortality with attempted PEA. In particular, in patients with a PVR disproportionately higher than the segmental obstruction visible by imaging, there is less benefit from PEA and a much higher risk of mortality. Some may be considered for lung transplantation instead. Decisions regarding operability in some patients depend on the combined clinical experience of the multidisciplinary team.
Operative risk is almost totally dependent on CTEPH, and concomitant procedures (CABG, valve replacement, etc) are performed as necessary without additional risk. The only comorbidity that may influence the decision to operate is severe parenchymal lung disease, but there are few absolute contraindications to PEA surgery and all patients should be referred after complete investigation for consideration of surgery. Age is not a barrier to surgery, and results in patients >80 years are good.
6.4.8.5.6 Surgical outcome
Overall the reported operative mortality for the PEA procedure is between 5–15% in experienced centres, but is dependent on the case mix of patients.
6.4.8.5.7 Medical management
While there is no doubt that most patients with CTEPH should undergo PEA, it is uncertain how to approach those with non-surgical distribution of the disease and those with residual PH post-PEA. Disease-targeted therapies for PAH are now being studied. Intravenous epoprostenol has been used as a bridge before surgery resulting in some haemodynamic stabilisation.227 ,228 Uncontrolled studies suggest a role for both sildenafil229 and bosentan.137 A randomised, placebo-controlled trial to establish safety and efficacy of bosentan is currently in progress. A proposed therapeutic approach to CTEPH is presented in fig 7.224 Current UK practice for medical treatment of CTEPH is to adopt the treatment algorithm as in IPAH until new evidence from randomised trials is available. Patients with significant postoperative PH or late recurrence of PH are considered for disease-targeted therapies.

6.4.8.5.8 Organisation of care
In the UK all patients with suspected CTEPH should be referred initially to their regional NCG designated PH centre who will then refer appropriate patients to the NCG designated centre for PEA surgery at Papworth Hospital.
RECOMMENDATION
62. All patients with CTEPH should be assessed for PEA unless they do not wish to undergo surgery.
63. An IVC filter should be inserted preoperatively.
64. Patients with distal CTEPH should be managed with disease-targeted therapies for PAH.
6.5 Patient-centred outcomes
Compared with healthy individuals, patients with PH suffer from limitations in their physical mobility, energy, emotional reactions and social isolation. Although measurement of HRQoL and QoL is important in this patient population, the impact of PH and its treatments on HRQoL or QoL has not traditionally been assessed in clinical practice.
In some clinical trials of PAH treatments HRQoL and generic measures such as the Short-Form-36, Minnesota, or the EuroQoL were assessed.11 ,114 ,116 ,127 ,172 ,230 The content of these is not disease-specific. A new disease-specific patient reported outcome instrument, CAMPHOR, has been developed.94 CAMPHOR correlates well with the most common surrogate end points used for assessing disease progression and is sensitive to small changes in health and QoL in this patient group.231
RECOMMENDATION
65. A QoL assessment such as CAMPHOR should be used in routine clinical practice.
6.5.1 Follow-up and shared care
Patients with PAH require life long follow-up since the drug treatments are not curative and the disease may break through drug treatment unpredictably. Routine follow-up should include a history and physical examination, 6MWT and CAMPHOR questionnaire. Further investigations should be performed according to the clinical status of the patient and include ECG, chest radiography, biomarkers, echocardiography, right heart catheterisation, cardiac magnetic resonance and CT.
Patients who live at a distance from a designated centre will be admitted to their local hospital in case of emergency. It is important that a local physician knows the patient. Depending on the interest of the physician and local hospital facilities shared care may be considered.
RECOMMENDATION
66. Patients with PAH, CTEPH and others treated with disease-targeted therapy should receive life-long follow-up in an NCG designated centre.
67. Patients taking disease-targeted therapy normally require 3 monthly hospital outpatient visits.
68. Patients living at a distance from a designated centre should also be followed up by a local physician.
6.6 Atrial septostomy
Patients with IPAH and a patent foramen ovale (PFO) have a survival advantage over those without a PFO,232 supporting the concept of atrial septostomy as a treatment for IPAH. The creation of an inter-atrial right to left shunt can decompress the right ventricle, increase left ventricular preload and increase cardiac output, improving systemic oxygen transport despite arterial oxygen desaturation.
The precise role of atrial septostomy in the treatment of IPAH remains uncertain, but evidence is suggestive of benefit in patients who are in WHO functional class IV with right heart failure refractory to medical therapy or with severe syncopal symptoms.233–235
The recommended technique is graded balloon dilation atrial septostomy which produces equivalent improvements in haemodynamics and symptoms but reduced risk compared with the original blade technique.235 ,236 A baseline mean right atrial pressure of >20 mm Hg and an oxygen saturation at rest of <80% breathing air indicates a high risk of procedure related mortality and in general atrial septostomy should be avoided. In UK practice atrial septostomy is considered only in patients who are failing medical therapies. This includes patients who are being considered for or awaiting transplantation.
Evidence suggests improvements in cardiac index following the procedure of 15–58% with improvements in 6MWT distance.235 ,237
Severe IPAH has been the main indication for atrial septostomy in adults, although other indications include PAH associated with surgically corrected congenital heart disease, distal CTEPH, PVOD, PCH and SSc APAH.
The impact of atrial septostomy on long term survival has not been established in prospective uncontrolled studies, but reports do support improvements in survival within the patient populations treated and late deaths are primarily due to progression of the pulmonary vascular disease which is unaffected by septostomy per se.235
RECOMMENDATIONS
69. Atrial septostomy should be regarded as a palliative or bridging procedure for patients with severe PAH failing on medical therapy.
70. Atrial septostomy should be performed by centres with experience in the procedure of graded balloon dilation atrial septostomy, the preferred choice of procedure.
71. Patients with advanced PAH whose right atrial pressure is >20 mm Hg and whose oxygen saturation at rest is <80% are at high risk of dying if the procedure is undertaken.
6.7 Transplantation
The advent of effective medical therapy for severe PAH has presently reduced the number of patients referred for transplantation. The long term outcomes of such patients remain uncertain and it is clear that lung or heart lung transplantation will remain an important mode of treatment both for patients failing medical treatment either following an initial period of clinical benefit or not. Transplantation guidelines have been published.238 Studies demonstrate up to 25% of patients with IPAH may fail to improve on disease-targeted therapy and the prognosis of those who remain in functional class III or IV remains poor.15 ,107
6.7.1 Prognostic factors in consideration of transplant listing
Aetiology
The prognosis of PAH varies according to its underlying aetiology. SSc APAH has a worse prognosis than IPAH even with prostanoids.112 ,239 Patients with PAH associated with congenital left to right shunts have an improved survival and this has been noted in patients with advanced disease on transplant waiting lists. PVOD and PCH have the worst prognosis due to the lack of disease specific medical therapy.
Functional studies
In addition to WHO functional class, exercise testing correlates with survival in IPAH. A 6MWT distance <332 m or a peak oxygen consumption <10.4 ml/min/kg is associated with a worse prognosis.54 ,59
Haemodynamics
Adverse predictors of poor survival include cardiac index <2 l/min/m2, right atrial pressure >20 mm Hg, a mean PAP >55 mm Hg and a mixed venous oxygen saturation <63%.1 It is important to note that the adverse haemodynamic data do not predict a lack of potential response to medical therapy.240
6.7.2 Choice of surgery
For patients with PAH, choosing the transplant operation is an important facet of preoperative evaluation. Both heart lung and isolated lung transplantations have been performed for pulmonary vascular disease but heart and lung transplantation should now be reserved for patients who are not candidates for lung transplantation alone. The threshold of unrecoverable right ventricular dysfunction remains unknown and severe dysfunction has been shown to be reversible after isolated lung transplantation. While afterload is immediately reduced by lung transplantation, right ventricular dysfunction does not improve immediately and haemodynamic instability is a common problem in the early postoperative period.241 Both single lung transplantation (SLT) and bilateral lung transplantation (BLT) have been performed for PAH. While recipient survival rates have been similar after SLT and BLT for PAH, any complication in the allograft following SLT is associated with severe hypoxaemia and there has been a move towards BLT. The International Society of Heart and Lung Transplantation (ISHLT) reported that in 2005 BLT was performed in 95% of isolated lung transplantations for PAH.
In PAH patients with Eisenmenger syndrome the option of HLT should be carefully considered, despite the fact both SLT and BLT have been combined with repair of cardiovascular anomalies.242
Patients with Eisenmenger syndrome due to ventricular septal defects have improved survival with HLT.243 Overall worldwide activity for HLT has dropped over the years with only 75 operations reported to the ISHLT in 2006.
Outcomes
Actuarial survival following transplantation for PAH has been well documented by the Registry of the ISHLT. The overall 5 year survival is 45–50%.244 Transplantation for PAH and Eisenmenger syndrome has the highest perioperative mortality among the major diagnostic categories of patients undergoing transplantation, and this is explained by the complexity of the surgery in severe PH.
RECOMMENDATION
72. Designated PH centres should establish a clear working relationship with a UK transplant centre to facilitate the referral process.
73. Patients should be referred if they fulfil the general international guidelines for transplantation and are functional class III or worse.
74. The potential need for transplantation should be discussed with all patients presenting in WHO functional class III. Patients in WHO functional class IV should be referred on presentation to a transplant unit if they appear to be potential candidates.
75. All candidates should be treated with disease-targeted therapy before undergoing transplantation. Transplantation should be undertaken when treatment with such therapy is failing.
76. Those patients who show significant improvements following medical therapy over the first 3 months and who in particular move to WHO class II functional status can defer further transplant assessment or listing.
6.8 New and future therapies
PAH research is currently thriving, with new drug treatments in clinical trials and several novel targets under active investigation. Interest in correcting endothelial dysfunction and increased vasomotor tone remains, but there is now greater emphasis on addressing directly the structural changes that accompany and in some cases precede the development of increased PVR. The pleotrophic effects of statins (anti-proliferation, pro-apoptosis, anti-inflammation) together with preclinical observations suggest this drug class may be beneficial in PAH, but this remains to be demonstrated. Tyrosine kinase inhibitors such as imatinib have attracted interest following a case report,245 but the results of formal clinical studies are needed to establish safety in PAH. Other interesting pharmacological strategies include inhibition of the serotonin reuptake transporter, activation of potassium channels, inhibition of Rho kinase, and modification of mitochondrial pyruvate dehydrogenase kinase. Cell based therapy is also under investigation. Endothelial progenitor cells from patients can be expanded in culture and there is interest in using these cells to deliver gene therapy to the pulmonary vascular bed. This is at a very early stage and further work needs to be done to establish the most appropriate cell phenotype to deploy.
PAH is a heterogenous condition and it is unlikely that one treatment protocol will suit all patients. Advances in our understanding of the genetic basis of FPAH have fuelled speculation that a specific corrective therapy may emerge for some patients. For many, effective treatment will lie with combination therapy. At present, the emerging strategy is to combine existing treatments (prostanoid, ERA and PDE 5 inhibitor) and new treatments will have to find their place in this hierarchy.
A challenge will be to demonstrate efficacy from novel therapies in clinical trials. Drugs which impact on vascular structure will not be expected to show acute effects on pulmonary haemodynamics. 6MWT distance has been widely used as an end point, although its limitations are widely appreciated. When looking at combination therapies and when recruiting patients with milder symptoms (WHO functional class I–II), significant increases in 6MWT distance may not be achieved due to a ceiling effect. It is clear other more sensitive tests will be required.
7. PULMONARY HYPERTENSION IN CHILDREN
7.1 Introduction
Pulmonary vascular disease may often progress while the lung is still developing and children are generally sicker than adults at presentation. The response to treatment is less predictable in children who need close monitoring and rapid escalation of therapy with any clinical deterioration. In IPAH the mean survival time without treatment is 10 months.
7.2 Causes of pulmonary hypertension in childhood
The Venice classification (box 1) includes all the types of PH encountered in childhood. The types of PH currently treated with disease-targeted therapy in children are shown in box 5.
Box 5: Types of pulmonary hypertension treated in children
1. Pulmonary arterial hypertension
1.1 IPAH
1.2 FPAH. (Children can express the same genetic mutations and deletions as adults with IPAH35)
1.3.1 Collagen vascular disease and the vasculitides
1.3.2 Postoperative congenital heart disease (not acute postoperative pulmonary hypertension, but that sustained over many years) which can behave like IPAH
1.3.2 Congenital heart disease which has never been operable, pulmonary vascular resistance having been elevated since birth plus the classical Eisenmenger syndrome
1.4.1 Pulmonary veno-occlusive disease
1.5 Persistent pulmonary hypertension of the newborn
2. Pulmonary hypertension with left heart disease
2.1 Left ventricular disease caused by cardiomyopathies and congenital left heart anomalies
3. Pulmonary hypertension associated with lung diseases
3.2 Interstitial and fibrotic lung disease
4. Thromboembolic disease is rare in childhood
7.3 Investigation of the pulmonary hypertensive child
It is mandatory to make an accurate and complete diagnosis before instituting treatment. There may be an informative history of fetal or neonatal incidents, premature delivery, intrauterine growth retardation, congenital diaphragmatic hernia, or chronic lung disease. Initial investigations include an ECG, chest x ray and a transthoracic echocardiogram. A 6MWT is performed if the child is able to cooperate. This is followed by a CT scan, and where appropriate a ventilation perfusion scan and a sleep study. Older children frequently require formal exercise testing and lung function tests. Evidence for CTD is sought. The definitive study is cardiac catheterisation following an established protocol with vasodilator testing using NO and a high inspired oxygen concentration.
7.4 Medical treatment of pulmonary hypertension in children
The treatment algorithm is a modification of the one designed for use in adults (fig 8) and has been developed from clinical experience. Children are sometimes too ill to undergo cardiac catheterisation246 and treatment may have to be instituted immediately.

{kind=link}
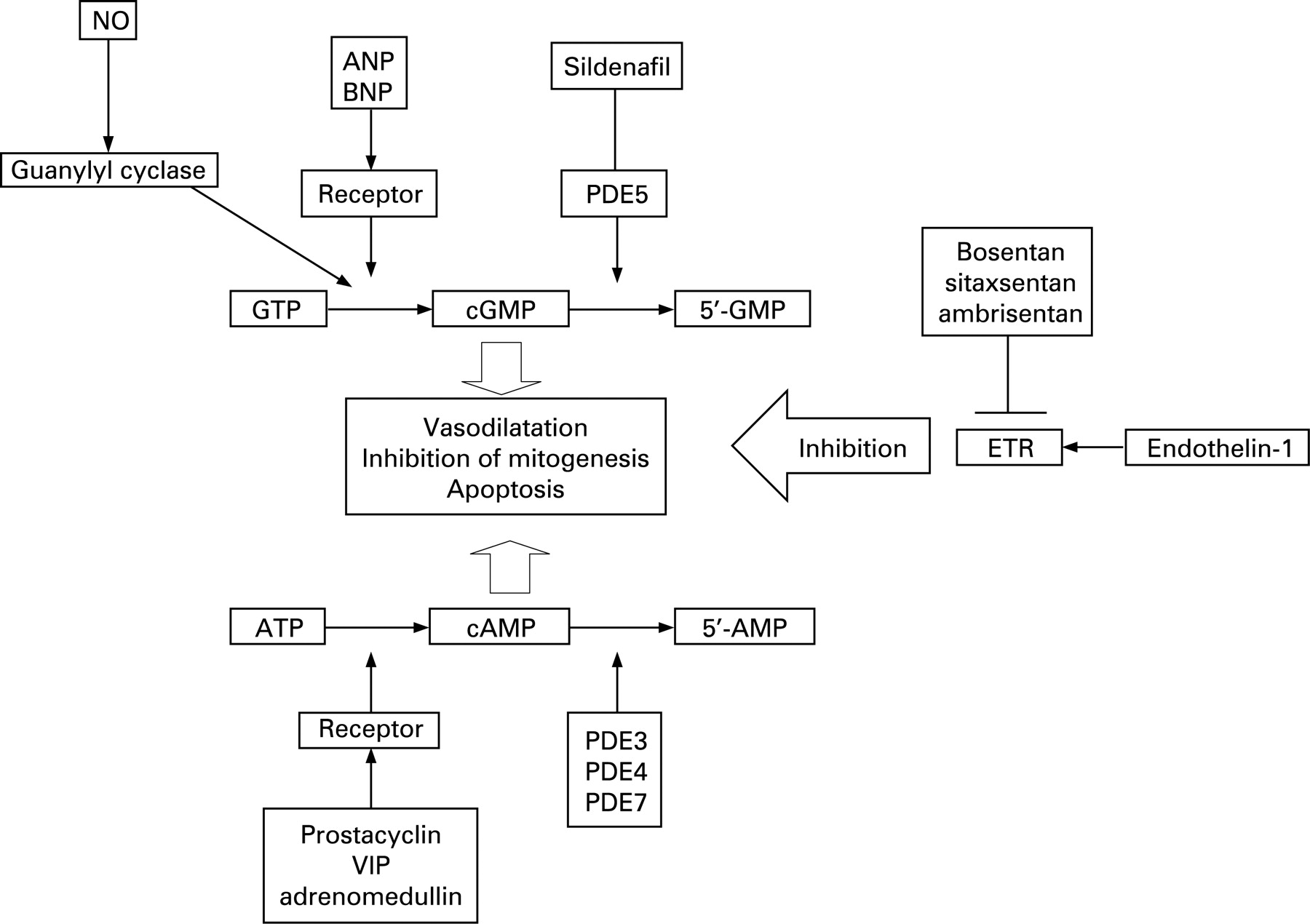{kind=link}
{kind=link}
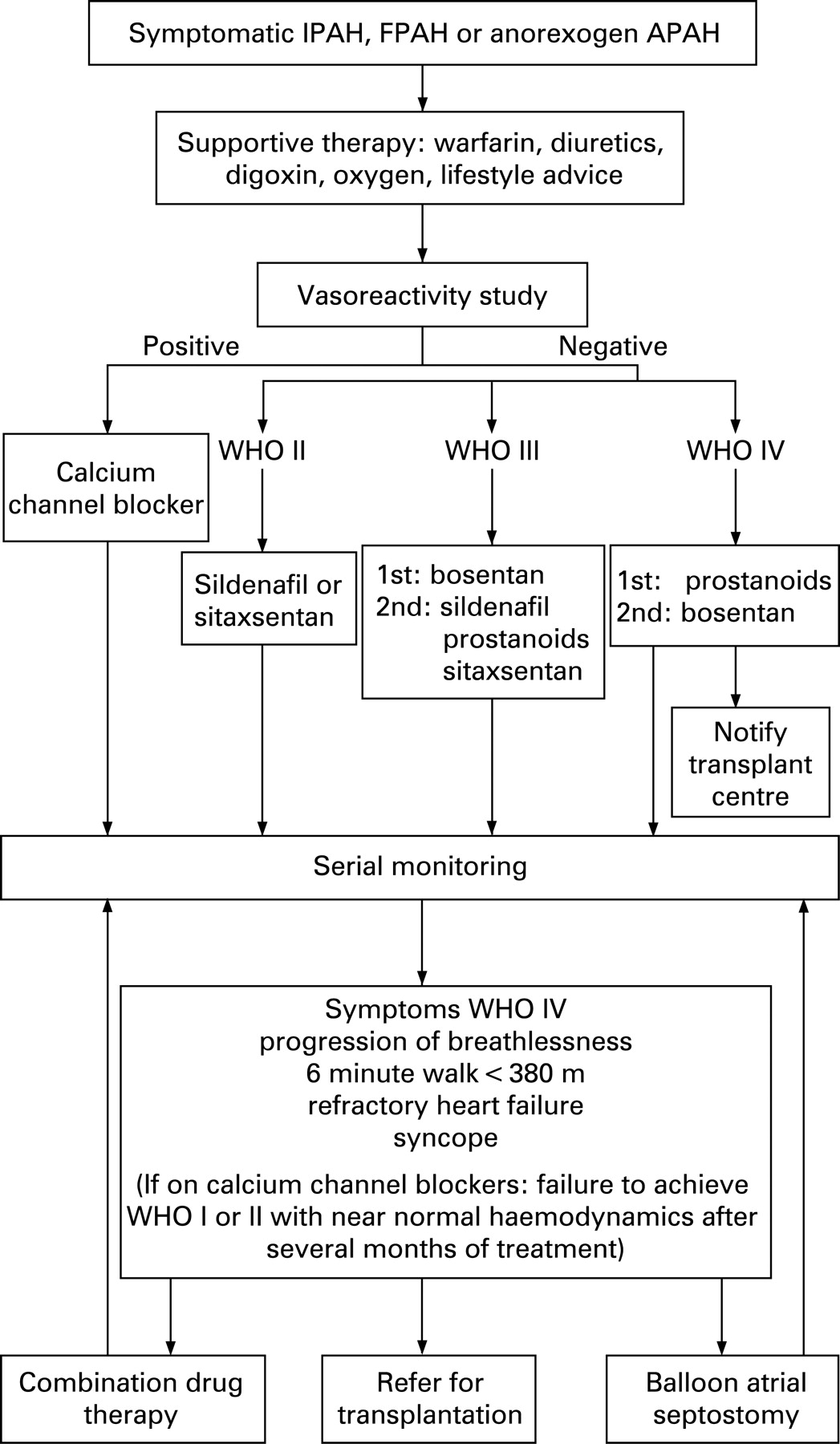{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Only 5% of UK patients have been positive responders to vasoreactivity studies. The majority of children are therefore treated with disease-targeted therapies. These include intravenous epoprostenol247 (the only drug for children tested in a placebo-controlled trial), other prostanoids, bosentan,248 and sildenafil.
Choice of therapy is determined by WHO functional class. The age of the child is important because several drugs used to treat adults are unsuitable for young children: subcutaneous treprostinil causes site pain which is poorly tolerated; small sick children cannot reliably inhale an adequate dose of iloprost at 2–3 h intervals. Combination therapy is often necessary.107 Treatment regimens vary because pathogenesis, age and understanding are variable. Future therapies for children await safety data from adult studies.
Children with severe PH and intact atrial and ventricular septa may have syncopal attacks which require an atrial septostomy.
7.5 Treatment outcome
7.5.1 Quality of life
Every effort is made to ensure that the children have as fulfilled a life as possible and to keep the children out of hospital as much as possible. Where possible those of nursery and school age go back to school at least for some time. This includes those receiving intravenous epoprostenol.
There is currently no disease-specific quality of life assessment for children. Therefore the SF10 (QualityMetric Incorporated, Boston, USA) is used. Parents and the older children complete the form at each outpatient visit.
The results show that although physical ability is limited to approximately 50% of normal, the psychological scores are 80–90% of normal at the first and subsequent visits.249 ,250 Data are similar for IPAH and APAH. The quality of life score does not relate to age, time since diagnosis, PVR, type of therapy (oral, intravenous, subcutaneous, or inhaled) or cause of PH.
7.5.2 Survival
Treatment with disease-targeted therapies for PAH improved survival in patients with severe pulmonary hypertension who were in WHO functional class III and IV at Great Ormond Street Hospital for Children (GOSHC). In children with IPAH the cumulative survival was 84% and 76% at 1 and 3 years, respectively.249 ,250 It was slightly better in those with APAH. These figures include children who died either at the onset of therapy or before therapy could be started.
IPAH
Survival is better in children given combination therapy than in those given monotherapy.249 ,250 According to the treatment algorithm, severely symptomatic children are given epoprostenol initially while the less symptomatic children are given bosentan. Children deteriorating on either therapy have the other drug added to their therapeutic regimen.
APAH
These children have benefited considerably from the medications used to treat patients with IPAH. The worst outcome was seen in children with postoperative pulmonary hypertension, despite being treated as aggressively as those with IPAH. Earlier recognition of pulmonary hypertension is essential in these children. Not surprisingly, the best outcome is seen in those with Eisenmenger syndrome.
Benefit of atrial septostomy
This procedure is only carried out on the severely symptomatic child with syncope, with or without right heart failure. More than 30 septostomies have been performed with no mortality. In a published series the mean age of the first 21 children treated was 8.4 years and the mean follow-up was 2.5 years.251 Atrial devices have been inserted in 10 children following which there has been no further syncope and all experienced a beneficial shift in WHO functional class.
Transplantation
Many children eventually fail medical treatment and require transplantation.
7.6 Current service provision and education
7.6.1 Organisation of the clinical network
The pulmonary hypertension team at GOSHC provides expert guidance and help to a network of paediatric cardiology centres at Leeds General Infirmary, Bristol Children’s Hospital, Southampton General Hospital, Freeman Hospital Newcastle upon Tyne, Birmingham Children’s Hospital, Yorkhill Hospital Glasgow and the Royal Hospital for Children, Belfast.
Each of these satellite centres has a paediatric cardiologist with a special interest in PH. The GOSHC team hold joint clinics with local teams every 2–3 months (except Belfast), and teleclinics are interposed as needed with Belfast and Glasgow. Welsh children are seen in the Bristol clinic, with a paediatric cardiologist from the Heath Hospital Cardiff.
A genetic counselling clinic is held at GOSHC for all UK children and their families. Parents frequently demand that their other children be screened for evidence of pulmonary hypertension.
Long term support in the community is essential. This is arranged by the Clinical Nurse Specialists who have established widespread links with paediatric community nurses throughout the UK and with several hospices for children.
RECOMMENDATIONS
77. The UK Children’s Service will accept referral of and/or be available to give advice for all children, even on suspicion of the diagnosis of PH.
78. All appropriate treatment modalities that are offered to adults should be available to children including intravenous epoprostenol.
79. Close liaison with community health care professionals and schools about the pulmonary hypertensive child is essential.
8. CLINICAL RESEARCH IMPLICATIONS
Current therapies for PAH slow down disease progression but are not curative. New approaches to treatment will be the only means to benefit patients. As the number of clinical trials expands in such a limited patient population there are already becoming too few patients to complete all the studies which include new indications for existing therapies, investigation of benefit in diagnostic subgroups, new therapies and combinations of therapies. This demand for clinical research can only be met if the maximum number of patients is entered into trials while refining techniques used for assessing therapeutic response including imaging and biomarkers.
RECOMMENDATION
80. Wherever possible patients with PH should be offered the opportunity to participate in clinical trials. This will be most efficiently achieved through designated centres which all have national and international collaborations.
Appendix 1: Clinical trials in progress
REFERENCES
Footnotes
Funding: The consensus meeting was sponsored by the patients’ association, PHA-UK. The publication costs of this document have been met by unrestricted educational grants from Actelion Pharmaceuticals, Encysive Pharmaceuticals, GlaxoSmithKline, LungRx, Pfizer, and Bayer Schering Pharma.
Competing interests: Iain Armstrong has received honoraria for lecturing and travel grants for conferences from Actelion, Schering and Encysive and is a member of the Nurse Advisory Board for Actelion. Dame Carol Black has been an advisor to Actelion, Cambridge Antibody Technology, Genzyme, and Merck & Co. Gerry Coghlan has received financial support from Actelion for nurse specialist, audit work, consultancy service, lecturing and support for congress attendance; Lilly-Icos for consultancy service; Pfizer for lecturing; Schering for nurse specialist financial support and consultancy services. Paul Corris has been a member of advisory boards for Actelion, Encysive and Pfizer and has received honoraria for lecturing from Actelion, Pfizer and Schering. Agnes Crozier is on the Nurse Advisory board for Actelion and has received travel grants for conferences from Pfizer and Actelion. Julia De Soyza has received travel grants from Actelion, Schering and Encysive and is a member of the Nurse Advisory Board for Actelion. Charlie Elliott has received honoraria for lectures from Actelion and travel grants from Actelion and Schering. Sean Gaine has been a member of advisory boards for Actelion, GlaxoSmithKline, Encycisive and Pfizer and has received honoraria for lecturing from Actelion, GlaxoSmithKline, Encysive, and Schering. The PH Unit has received contributions to research funds from Pfizer, Schering, Actelion and GlaxoSmithKline. Michael A Gatzoulis has received honoraria for lecturing and advisory board meetings and unrestricted educational grants from Actelion and Pfizer. Simon Gibbs has been a member of advisory boards for Actelion, GlaxoSmithKline, Pfizer and Encysive and has received honoraria for lecturing from Actelion, GlaxoSmithKline and Schering. Wendy Gin-Sing is a member of the nurse advisory board for Actelion and has received grants to attend conferences from Actelion, Pfizer, Schering, Encysive, United Therapeutics and GlaxoSmithKline, and honoraria for lecturing from Actelion, Encysive and United Therapeutics. Clive Handler has received travel grants for conferences from Actelion and Encysive. Luke Howard has received travel grants and lecture fees from Actelion and GlaxoSmithKline. Sheila G Haworth has received consulting fees from Actelion, and accepting honoraria from Pfizer. The PH unit has received support for the clinical service from Actelion and GSK. Rodney Hughes has received honoraria for lecturing and travel grants from Actelion, Schering and United Therapeutics. Martin Johnson has received travel grants for conferences from Actelion, GlaxoSmithKline and Encysive. David G Kiely is a member of advisory boards for Actelion, Pfizer and Encysive Pharmaceuticals and has received honoraria for lecturing from Actelion, Schering, Pfizer and United Therapeutics. Jim Lordan has been a member of an advisory board for Encysive and has received travel grants and honoraria from Encysive. Nicholas Morrell has received honoraria for educational lectures from Actelion, United Therapeutics and GlaxoSmithKline, and has received research funding from Actelion and Novartis. Andrew Peacock is on the advisory boards for Actelion, Pfizer, GSK and Encysive, and has received lecture fees from Actelion, Pfizer, GSK and Encysive. Joanna Pepke-Zaba is a member of advisory board for Actelion and Pfizer, and has received honoraria for lecturing from Actelion and Schering. Karen Sheares has received travel grants for educational meetings/conferences from Actelion, GlaxoSmithKline and United Therapeutics. Martin Wilkins has received contributions to research funds from Pfizer and Actelion and honoraria from Pfizer, Actelion, GlaxoSmithKline and Encysive. John Wort has received contributions for research funds and travel grants from Actelion.