Article Text

Statistics from Altmetric.com
- pulmonary hypertension
- primary pulmonary hypertension
- persistent pulmonary hypertension of the newborn
- pulmonary vascular obstructive disease
- nitric oxide
Severe, sustained pulmonary hypertension leads to pulmonary vascular obstructive disease, which is potentially fatal. But recent advances in genetics and cell biology provide insights into the pathogenesis of this disease and new treatments offer an improved quality of life and increased survival. The field is moving rapidly, and it is time to adopt a more positive and aggressive approach to the management of pulmonary hypertension in children. The sustained clinical and haemodynamic improvement seen in many adults with primary pulmonary hypertension treated with continuous prostacyclin, and data from numerous experimental studies indicate that it is possible to arrest and perhaps even reverse the disease process. Potential is likely to be greater in the young in whom the vasculature is still remodelling. However, pulmonary hypertension is frequently unrecognised in early childhood and most children are referred late in the course of the disease, emphasising the need to increase awareness of the condition. This review focuses on the more common forms of pulmonary arterial hypertension in the young— persistent pulmonary hypertension of the newborn (PPHN), primary pulmonary hypertension (PPH), and pulmonary hypertension associated with congenital heart disease. It attempts to represent our current understanding of pulmonary hypertension in childhood, highlighting the key features of the condition, and offers a glimpse into the future.
DIAGNOSTIC DEFINITIONS OF PULMONARY HYPERTENSION
Pulmonary hypertension is defined as a pulmonary arterial pressure > 25 mm Hg at rest or > 30 mm Hg on exercise, although pulmonary hypertension in childhood is usually associated with considerably higher pressures. Pulmonary hypertension can be described as either primary, being of unknown aetiology, or secondary resulting from cardiac or parenchymal lung disease. This description is unsatisfactory, however, since it takes no account of the similarities in pathobiology and response to treatment between primary and certain other types of pulmonary hypertension. It narrows our perspective. A new classification was proposed at a World Health Organization symposium in 1998, based on anatomy, clinical features, and an appreciation of the commonality of at least some of the underlying mechanisms.1 PPH and pulmonary hypertension related to congenital heart disease, PPHN, connective tissue disease, HIV infection, drugs, and toxins were grouped together as “pulmonary arterial hypertension”. This new classification encourages the extension of therapeutic modalities known to be effective in PPH to other forms of hypertension, in both adults and children.
Further clarification is necessary in children with congenital heart disease. In the majority of children pulmonary arterial hypertension is usually caused and driven by a cardiac abnormality which leads to the development of the Eisenmenger syndrome. However, in some children the abnormality is, and always has been, haemodynamically insignificant. Clinically these children behave as though they have PPH, and can be treated as such.
PATHOGENESIS OF PULMONARY VASCULAR DISEASE IN CHILDHOOD
During the past few years we have gained considerable insight into the molecular mechanisms responsible for the development of pulmonary vascular disease. Findings can be related to current treatment strategies and may suggest a new therapeutic approach. Genetic studies have concentrated on familial PPH (FPPH) and the mutations recently identified in FPPH have not yet been sought systematically in other forms of pulmonary hypertension.
Persistent pulmonary hypertension of the newborn
Failure of the pulmonary circulation to adapt normally to extrauterine life causes PPHN, a condition with a high morbidity and mortality despite the advent of inhaled nitric oxide (NO) therapy. PPHN is multifactorial in origin, although commonly associated with congenital and acquired hypoxic lung disease and congenital heart defects. Rarely, it is idiopathic. Irrespective of aetiology, during the first few days of life the intrapulmonary arterial wall structure is similar to that seen in fetal life and neonatal remodelling is impaired2 (fig1). Functional studies demonstrate impairment of the NO pathway, sometimes a deficiency of the NO substrate L-arginine, increased concentrations of the endogenous inhibitor asymmetric dimethyl arginine, and persistently high circulating endothelin concentrations. Studies on normal animals reveal low NOS activity and relatively poor endothelial dependant relaxation at birth, and these systems fail to mature properly in PPHN.3 Different relaxation pathways (NO, prostaglandin, endothelium derived hyperpolarising factor) mature at different rates and have different vulnerabilities to insult. Vasoconstrictor endothelin ET-A receptors increase and endothelial vasodilator ET-B receptors decrease. Thus failure to reduce the pulmonary vascular resistance after birth appears to involve a primary structural abnormality, failure of endothelial dependent ± independent relaxation, and an excess of vasoconstrictor activity(s). The rationale for giving NO and phosphodiesterase inhibitors is that there is an absolute or relative lack of the endogenous substance. Oxygenation usually improves and the need for extracorporeal membrane oxygenation is reduced. New strategies directed at antagonising vasoconstriction and modifying smooth muscle cell cytoskeletal remodelling are indicated. Outcome depends on causality. Some babies who appeared to recover normally were later found to have persistent arterial medial hypertrophy.
The relation between PPHN and PPH is uncertain but children who present with PPH during the first years of life frequently have a history which suggests that they were pulmonary hypertensive from birth. A history of prenatal stress and perinatal morbidity is not uncommon. Occasionally an infant thought to have PPH is found to have a secundum atrial septal defect. This is usually an incidental, though protective abnormality, and should not be closed.
Primary pulmonary hypertension
The number of children diagnosed with PPH and being referred for treatment has increased greatly since it became apparent that even infants could benefit from the treatments given to adult patients with the disease.
Genetics
Familial PPH
Only 6% of cases of PPH have been reported as familial.
The disease is transmitted as an autosomal dominant trait, with incomplete penetrance. The chance of a person carrying a gene for FPPH developing the disease is higher in females (0.30) than males (0.15), and a female predominance is present from early childhood. FPPH shows gene anticipation, the disease frequently presenting at an earlier age in successive generations.
The FPPH locus maps to chromosome 2q31-32 and germline mutations have been identified in the bone morphogenetic protein receptor -II (BMPR2).4 The BMPs form the largest group within the transforming growth factor β (TGF-β) family of cytokines. The PPH disease associated mutations identified would be expected to disrupt signalling pathways mediated by BMPR2, thereby removing a mechanism for keeping vascular remodelling in check and facilitating abnormal proliferation of pulmonary vascular cells.
Mutation in another TGF-β family member, the type I receptor gene, activin receptor-like kinase (ALK1) has been implicated in hereditary haemorrhagic telangectasia associated pulmonary hypertension.5
It is likely that other PPH genes remain to be identified. BMPR2 mutations have not been identified in some 60% of FPPH cases, and most sporadic cases do not appear to harbour BMPR2 mutations. There are also families in whom pulmonary hypertension is associated with haemoglobinophathies and platelet storage defects.
Sporadic PPH
BMPR2 defects have been described in 26% of sporadic cases of PPH, in some cases arising as “de novo” or spontaneous mutations.6 Sporadic and familial PPH have the same pathological features and the patients can be treated in the same manner.
Pathobiology
At necropsy, most adults have advanced pulmonary vascular obstructive disease with plexiform lesions (fig 2⇓). This picture is also common in older children and can be seen before three years of age. Particularly in young children, however, the cellular changes can be restricted to severe pulmonary arterial medial hypertrophy with pronounced intimal proliferation, lesions which are more likely to be potentially reversible. The instigators of this process are uncertain. Loss of one normal BMPR2 allele does not, in itself, produce the phenotype. It is now thought that PPH affects those with a genetic predisposition to respond adversely to a variety of stimuli, and that the clinical and structural findings represent the final common pathway. The following are thought important in the pathogenesis.

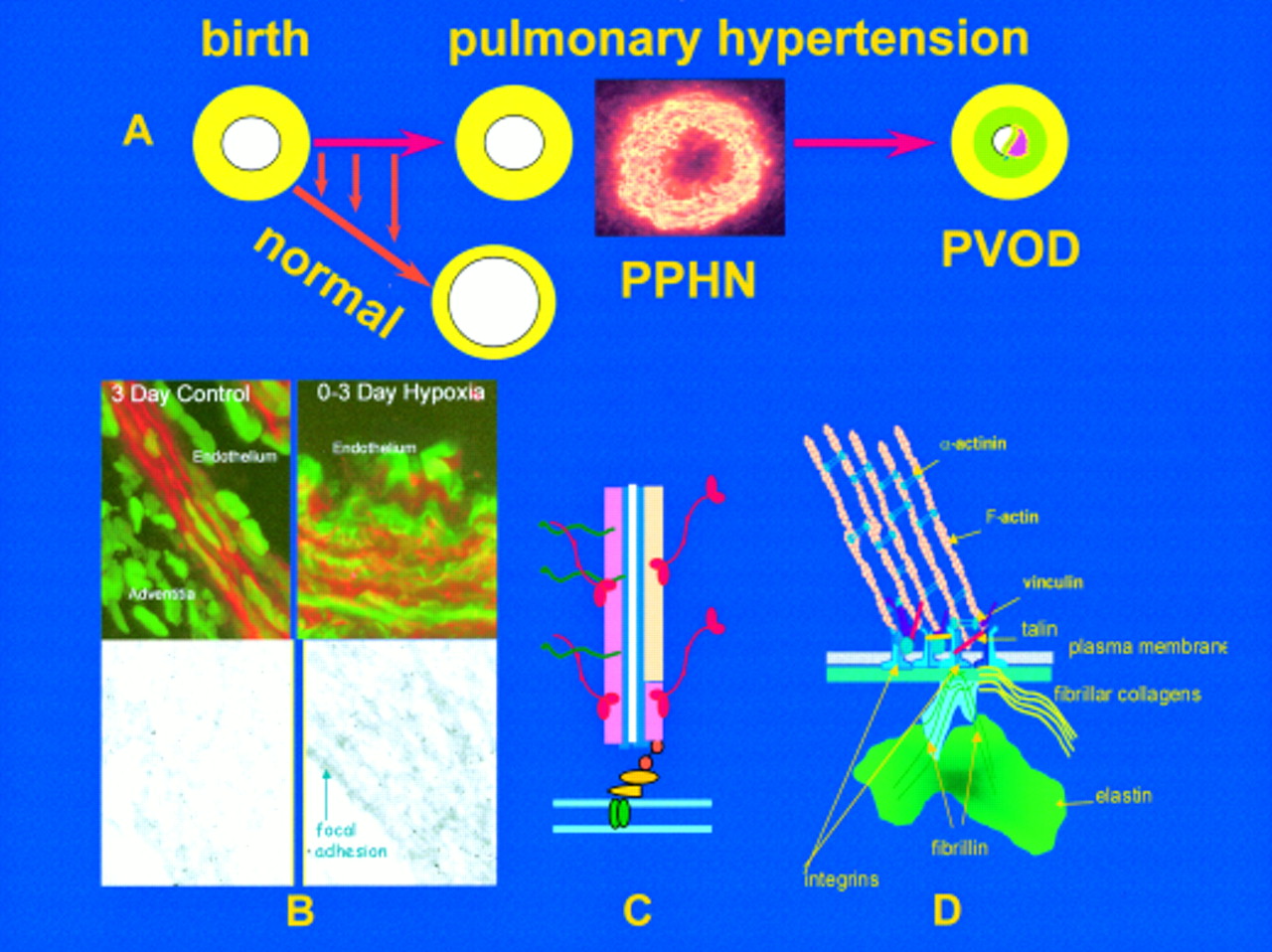
The upper figure (A) illustrates the rapid reduction in pulmonary arterial wall thickness occurring immediately after birth in the normal lung. This process is profoundly disturbed in persistent pulmonary hypertension of the newborn (PPHN) and an increase in medial thickness eventually leads to pulmonary vascular obstructive disease (PVOD) if the pressure remains high. Insert shows abnormal, hypertensive human peripheral pulmonary artery at three days, stained for γ actin. Mechanisms are illustrated in B, C, and D. (B) Confocal and transmission electron microscopy shows, in the left hand panel, the normal porcine peripheral pulmonary artery, and in the right hand panel, the pulmonary hypertensive vessel at three days. Normal remodelling entails reorganisation of the smooth muscle cell actin cytoskeleton which undergoes transient disassembly as the cells thin and elongate to spread around an enlarging lumen. In PPHN larger cells are packed with red actin myofilaments (phalloidin stained) while immuno-electron microcopy reveals sheets of actin bundles labelled with gold particles rather than actin being diffusely distributed in the cytosol. (C) Illustration of a myofilament. (D) All aspects of vessel wall remodelling are disturbed in PPHN, from the excessive myofilament assembly within the cell, focal adhesion remodelling at the membrane, and excessive connective tissue deposition in the matrix. Gene expression of tropoelastin and type I procollagen is abnormally high and steady state protein concentrations are increased. Vessels appear to become fixed in an incompletely dilated state. This is the beginning of PVOD in the young if the process cannot be arrested therapeutically.

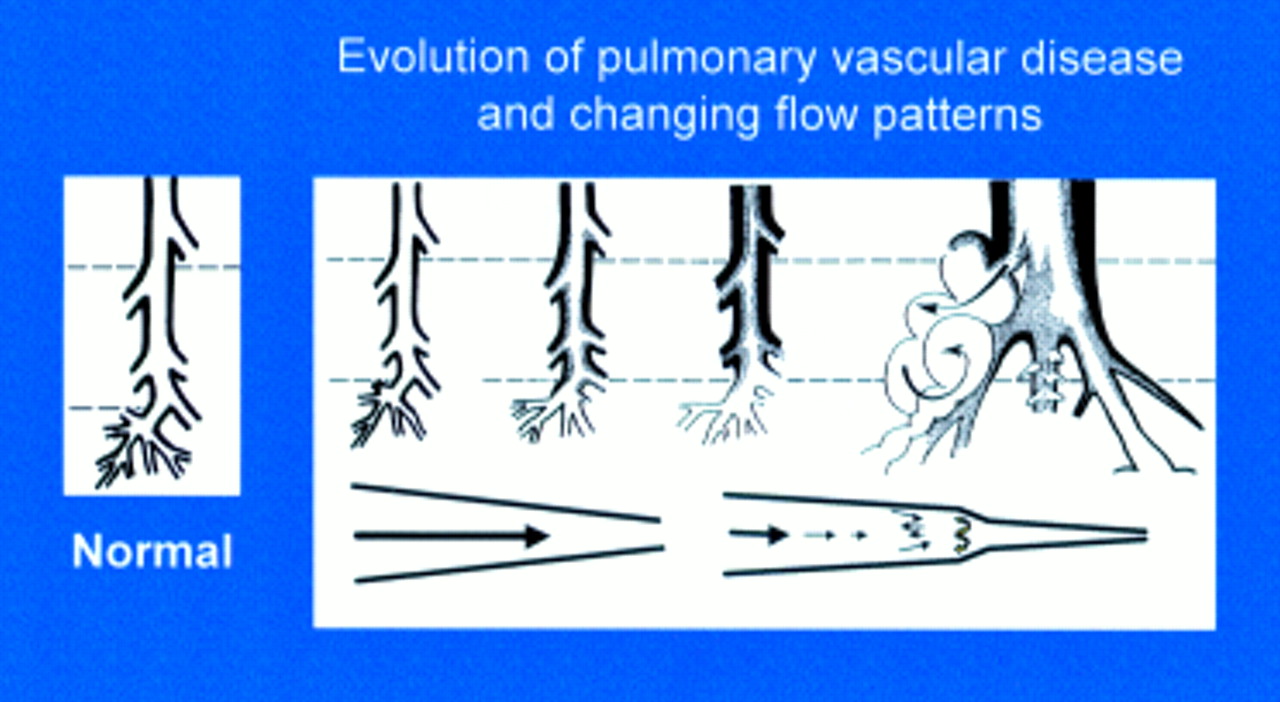
Evolution of pulmonary vascular disease in the young showing the early reduction in number of arteries, the increase in muscularity, and development of intimal proliferation. Two classification systems are used to describe these changes, the more recent concentrating on the early structural changes preceding intimal damage in a pulmonary vascular bed which is still developing, the other being the Heath and Edwards classification. A biopsy must be taken at sufficient depth (upper interrupted line) to include some pre-acinar arteries in which intimal proliferation first develops. For a limited time the more peripheral arteries can have a misleading near-normal appearance, a predilatation phase associated with an increase in mortality and morbidity at and after intracardiac repair. Blood flow patterns change from laminar to turbulent with disease progression.
Endothelial dysfunction
Concentrations of circulating endothelin, a powerful vasoconstrictor and mitogen, are raised and expression of endothelin converting enzyme is increased. Prostacyclin and NO, vasodilators with antiproliferative and antimigratory properties, are reduced. Chronic treatment with prostacyclin or one of its analogues is a proven, effective treatment7 while inhaled NO, NO donors and the phosphodiesterase 5/6 inhibitor sildenafil are as yet unproven alternatives/adjuncts in terms of both efficacy and safety. Endothelin receptor antagonists have proved safe and effective in small trials, mostly in adult patients, and are being trialled in younger patients.8
Intense vasoconstriction
Intense vasoconstriction is thought to be an early, common response to injury. Although pulmonary vascular disease is usually well advanced at presentation, the pulmonary vascular resistance falls in many patients on acute vasodilator testing, more often in children (50–60%) than in adults (20%). Calcium channel blockers are an accepted, conventional treatment, known to prolong survival in adults. Both hypoxia and anorexic agents which can cause pulmonary hypertension inhibit potassium currents in pulmonary artery smooth muscle cells causing membrane depolarisation, which promotes an increase in intracellular calcium concentration and hence vasoconstriction.9 Moreover, finding dysfunctional voltage gated potassium channels in primary but not secondary pulmonary hypertension suggests that potassium channels may play a significant role in the pathogenesis of PPH. Indeed, there is a general downregulation of potassium channel function in proliferating human pulmonary arterial smooth muscle cells.10 In terms of potential treatment, ATP sensitive potassium channel openers probably offer the greatest promise since these agents are potent dilators of the pulmonary circulation and are still able to promote membrane hyperpolarisation in proliferating smooth muscle cells. However, the recent evidence that non-voltage dependent calcium entry pathways (transient receptor potential channels) are upregulated during pulmonary vascular cell growth, could point to other novel targets in the treatment of this disease.
Platelet function
There is an excess of thromboxane in relation to prostacyclin, predisposing to vasoconstriction and platelet aggregation. The role of serotonin in the pathogenesis of PPH is still uncertain, but raised plasma concentrations and impaired platelet storage of serotonin can occur. Serotonin transporters are overexpressed on pulmonary arterial smooth muscle cells.11 Raised fibrinopeptide A concentrations and pathological studies indicate thrombosis in situ, and there is evidence of impaired local fibrinolysis. Anticoagulation increases survival in adults and is used routinely in children.
Dysfunction of the immune system
PPH appears to be autoimmune in some children, and pulmonary hypertension is a component of several autoimmune disorders, particularly scleroderma.
Whatever the nature of the initiating stimulus, the onset of pulmonary vascular disease is characterised by abnormal proliferation of endothelial and smooth muscle cells and fibroblasts (fig. 3⇓). Identifying a mutation in the BMPR2 receptor implicating defective control of vascular remodelling puts the structural abnormalities back in the forefront of research interest as being a/the prime mover in the pathogenesis, rather than being viewed always as the inevitable consequence of endothelial injury. Monoclonal cell expansion is thought to lead to the production of plexiform lesions in a subset of adult patients with PPH.
{kind=link}
{kind=link}
{kind=link}
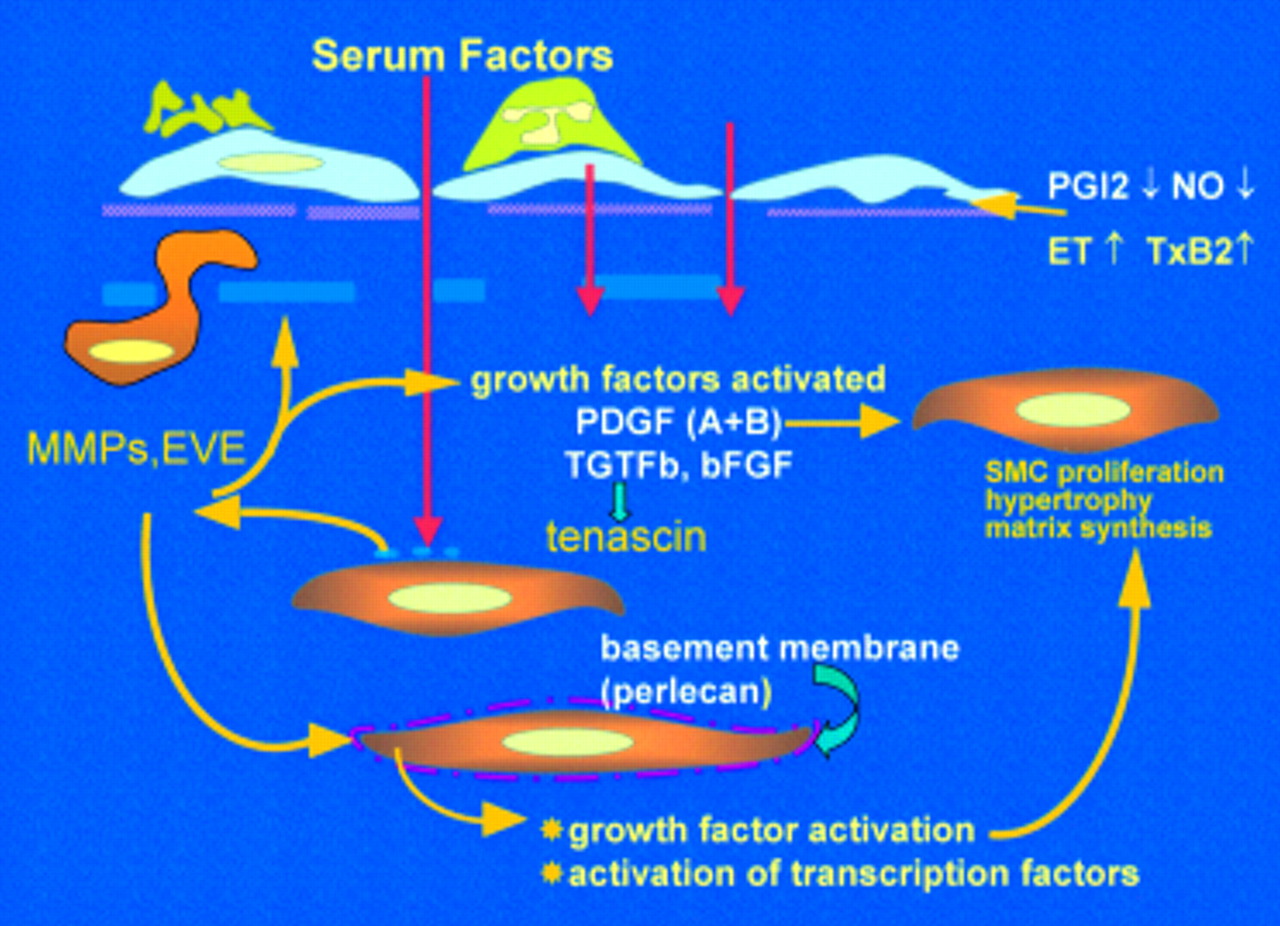
Factors driving the evolution of pulmonary vascular obstructive disease. Endothelial dysfunction reduces release of prostacyclin and nitric oxide (NO), causes adherence of activated platelets and leucocytes, enhanced release of thromboxane and endothelin, and loss of barrier function with leakage of serum factor into the subendothelium. This last action is thought to heighten activity of metalloproteinases (MMPs), including the proteolytic enzyme endogenous vascular elastase (EVE)14,21 released from smooth muscle cells, to help induce structural remodelling, cause smooth muscle cell activation, disruption of the internal elastic lamina, and facilitate smooth muscle cell migration. MMPs also activate growth factors normally sequestered in the matrix in an inactive form. Increased tenascin expression is associated with cell proliferation, its downregulation with apoptosis. Tenascin amplifies the proliferative response to epidermal growth factor and fibroblast growth factor (FGF)-2 in vitro. Expression of fibronectin is widespread and this glycoprotein can facilitate smooth muscle migration. The innermost smooth muscle cells cease to express many smooth muscle specific contractile and cytoskeletal proteins before migrating through gaps in the internal elastic lamina. Changes in phenotype are widespread.
Congenital heart disease
Intrauterine pulmonary vascular disease is unusual, and the disease generally starts at birth.12 The rate of change depends on the type of intracardiac abnormality, but some exceptional children appear to be genetically predisposed to develop an accelerated form of the disease. Endothelial cell damage, medial smooth muscle cell hyperplasia, hypertrophy, and site specific changes in cell phenotype are well described in early infancy12 (fig 2⇑). Respiratory unit arteries, about half of which normally form after birth, are reduced in size and number. This is the morphological substrate of pulmonary hypertensive crises, which most often occur in the presence of potentially reversible structural abnormalities. Endothelial dysfunction is present early. In potentially operable children the relaxation response to acetylcholine is impaired, basal NO production may be raised initially but then decreases, and the ratio of thromboxane to prostacyclin is raised, tipping the balance in favour of vasoconstriction and platelet aggregation. Impaired endothelial dependant relaxation occurs later in association with elevation in resistance and more advanced structural disease (fig 3⇑). Dilatation and plexiform lesions contain abundant vascular endothelial growth factor (VEGF) which co-localises with TGFβ1. VEGF induces endothelium dependent relaxation, which may help ensure continued perfusion of the capillary bed. But it is also a potent angiogenic factor, and TGFβ upregulates its angiogenic activity in vitro. The VEGF in the plexiform lesions could in theory stimulate angiogenesis. As intimal obstruction develops, flow becomes more turbulent (fig 2⇑) and in vitro studies suggest that this is likely to have an unfavourable influence on gene transcription. Laminar flow is associated with activation of genes such as eNOS and cyclo-oxygenase COX2 but turbulent flow is associated with the localised upregulation of VCAM-1 and ICAM-1, encouraging leucocyte recruitment and activation.13 Changes in mechanical stress also alter expression of specific genes in the smooth muscle cell, such as platelet drived growth factor.
Abbreviations
ALK1: activin receptor-like kinase
BMPR2: bone morphogenetic protein receptor-II
FPPH: familial primary pulmonary hypertension
NO: nitric oxide
PPH: primary pulmonary hypertension
PPHN: persistent pulmonary hypertension of the newborn
PVOD: pulmonary vascular obstructive disease
TGF-β: transforming growth factor β
VEGF: vascular endothelial growth factor
Postoperative pulmonary hypertension
The patient with repaired congenital heart disease has effectively been turned into a patient with PPH, with the added problem of a compromised myocardium. Survival is significantly worse in the untreated patient with PPH than in most patients with Eisenmenger syndrome. Assuming that these patients cannot be helped by further surgery, they should generally be treated as though they had PPH, and without delay.
Pulmonary hypertension and portal hypertension
An increased pulmonary arterial pressure can sometimes complicate the question of liver transplantation and necessitate careful haemodynamic assessment. The pathological features can resemble those found in hypertensive congenital heart disease, possibly caused by vasoconstriction because the damaged liver cannot degrade circulating vasoconstrictor mediator(s). But generalised pulmonary arterial dilatation can also occur. Pulmonary hypertension is not usually a contraindication to liver transplantation.
Pathobiology: a comment
Clinical and experimental studies have identified potentially important structural and functional abnormalities, but whether these are a cause or a consequence of the disease remains to be determined. Experimental models of PPHN have demonstrated that the term “endothelial dysfunction” does not apply to all aspects of endothelial function, but to specific signal transduction pathways in certain segments/regions of the pulmonary vascular bed. Functioning pathways should be identified and exploited. Strategies shown to be effective in attenuating the hypertensive response to hypoxia and monocrotaline in rats include endothelin receptor blockers, modulating potassium channels, inhibition of 5-lipoxygenase activating protein, serine elastase inhibitors,14 inhaled NO, and inhibition of 3′5′guanosinemonophosphate specific phosphodiesterase. In vitro studies indicate that there will be a role for smooth muscle growth inhibitors. The approach to gene therapy has concentrated on the overexpression of vasodilator genes, principally NO and prostaglandin I synthase, and results are encouraging.15,16
Tackling the different facets of angiogenesis is problematic. Growth of new vessels is a priority in the young who have developed pulmonary hypertension before the lung fulfilled its growth potential. Intratracheal (VGEF) 165 gene injection attenuated hypoxic pulmonary hypertension in rats, but the mechanism is uncertain. VEGF may not stimulate growth of normal vessels and in man it is abundant in plexiform lesions, which some have described as a form of uncontrolled angiogenesis. Evidence that advanced disease can be arrested has come from clinical experience with continuous intravenous prostacyclin therapy in PPH.17 Prostacyclin appears to be acting primarily by structurally remodelling the pulmonary vasculature rather than acting solely as a pulmonary vasodilator. New treatments will preferentially target the long term control of vascular remodelling rather than vasoconstriction. In young people, the aim must be to act as quickly and as effectively as possible to re-track pulmonary arterial remodelling along the normal pathway.
DIAGNOSIS AND CLINICAL INVESTIGATION OF PPH
In PPH, symptoms vary and are age related. Infants and young children may fail to thrive, tire easily, have exertional dyspneoa, and occasionally have chest pain. Symptoms suggestive of pulmonary hypertensive crises as well as syncope can occur at any age. In patients treated with chronic vasodilator therapy the most important determinants of survival are: (1) age—a 5 year survival of 88% in children of less than 6 years of age, as compared with 25% for older children; and (2) the acute response to prostacyclin—the 5 year survival being 86% as compared with 33% for non-responders. In congenital heart disease, symptoms and signs reflect the natural history of pulmonary vascular disease with and without surgery in the different anomalies.
The accepted approach to evaluating a child who has, or is suspected of having pulmonary hypertension is well described18. The following tests are crucial:
Cross sectional echocardiography clarifies intracardiac anatomy and an atrial communication is sought, particularly in the presence of an anatomically normal heart. Estimation of the pulmonary arterial pressure, right atrial and ventricular cavity size, and ventricular function is essential, and posterior bowing of the interventricular septum is sought in the presence of normal intracardiac anatomy.
At this point children should be referred to a specialist centre for further evaluation and treatment.
Exercise test, a six minute walk test or surrogate according to age and capacity, to measure the degree of functional impairment. In PPH exercise capacity correlates with right atrial pressure, pulmonary arterial pressure, and cardiac index. Pronounced limitation (< 10% predicted) is associated with increased risk at cardiac catheterisation.
Pulmonary function tests, which reflect clinical severity and haemodynamic status.
Oxygen saturation measurements, including a sleep assessment.
All the above tests are also used to monitor progress and the response to treatment. Tests carried out primarily to detect chronic thromboembolic disease are rarely indicated in childhood.
Cardiac catheterisation
Adequate sedation and meticulous attention to acid base status and blood loss is essential in children. Following a conventional study, acute vasodilator testing is carried out using 100% oxygen and short acting vasodilators such as inhaled NO, intravenous epoprostenol, and intravenous adenosine. Patients are perhaps best studied under light general anaesthesia, with respiratory gas analysis. Using measured oxygen consumption together with arteriovenous oxygen difference (bound and dissolved) cardiac output is calculated and pulmonary vascular resistance determined. In children who are extremely ill, cardiac catheterisation should not be unduly prolonged but testing with at least 100% oxygen and inhaled NO is mandatory in all cases because long term treatment is based on the response. A positive response to acute vasodilator testing is defined as ≥ 20% fall in pulmonary arterial pressure in the presence of an unchanged or increased cardiac output. Atrial septostomy/septectomy should be considered at the time of diagnostic catheterisation in severely ill children (particularly if there is a history of drop attacks) whose anatomy is such that there is no opportunity for right to left shunting to acutely decompress the right heart and improve systemic output.
An open lung biopsy may be indicated in complex congenital heart disease, suspected veno-occlusive disease, and vasculitis. Assessment includes quantitative morphometry to determine vascular development and a description of the pathological abnormalities. When evaluating an individual lung biopsy it is wiser to describe all the abnormalities present rather than to try to classify them, and relate the structural findings to the clinical and haemodynamic data. In young children with congenital heart disease the Heath and Edwards classification can underestimate the risk of surgery when severe medial hypertrophy (grade I) and/or exuberant cellular intimal proliferation (grade II) can be associated with pulmonary hypertensive crises and a high resistance.
MANAGEMENT OF THE PULMONARY HYPERTENSIVE CHILD
Lessons learnt in the management of children with PPH are now being applied to other forms of pulmonary hypertension in childhood.
Primary pulmonary hypertension
Treatment for PPH is treatment for life. The therapeutic regimen has to be tailored to meet the needs of each individual and adjusted as and when required according to changes in clinical and haemodynamic status. Children need close monitoring of the clinical course to ensure that a satisfactory response to treatment is sustained, with recatheterisation if necessary. Optimising the management of these patients greatly improves quality of life and survival.
Children with a positive response to acute vasodilator testing are given calcium channel blockers, usually nifedipine. Actuarial survival increased in adults treated with this drug. However, the magnitude of response which predicts long term survival is unknown in the young and clinical deterioration demands urgent revision of treatment.
Children unresponsive to acute vasodilator testing are not treated with calcium channel blockers which can have adverse effects and precipitate or worsen right heart failure. Older, compliant children in New York Heart Association functional class II can take nebulised iloprost which has a similar molecular structure to epoprostenol. But regular, effective dosing (6–12 times a day) is difficult in young children. The dual endothelin receptor antagonist Tracleer (bosentan), efficacious in adults, is now being trialled in children. The oral prostacyclin analogue beraprost sodium is efficacious in adults, but recommended only for those with less severe pulmonary hypertension and is largely untried in children. The subcutaneous analogue of prostacyclin treponistil (UT-15) is too painful for use in children. The phosphodiesterase inhibitor sildenafil is untrialled, its effect appears to be relatively short lived in sick children, and there is a risk of irreversible retinal damage linked to phosphodiesterase VI inhibition. The proven treatment of choice for the very sick child is chronic intravenous epoprostenol (prostacyclin) therapy. The dose is titrated according to clinical response, subjective and objective. Children generally need much higher doses of prostacyclin than adults and can become very tolerant of the drug, requiring constant, aggressive, upward adjustment of their dosage. Despite the obvious logistical problems, infants and young children can be managed satisfactorily. Training of two family carers by experienced nursing staff and a network of local support is essential. The side effects of the drug experienced by children are similar to those seen in adults. Clinical and haemodynamic improvement is generally sustained.
Other management strategies include:
supplemental domiciliary oxygen—this provides symptomatic improvement for those with systemic arterial desaturation
anticoagulation—warfarin rather than aspirin or dipyridamole is recommended to prevent thrombosis in situ, although aspirin is more tolerable in early infancy
supportive medical therapy—diuretics are indicated to control the fluid retention of right heart failure, but fluid can also appear with the onset of medical therapy; there is no good evidence that digoxin is helpful in the treatment of right heart failure in children
atrial septostomy/septectomy, if indicated
organisation of care in the community and contact with patients’ support groups is essential.
Screening and genetic testing
Parents almost invariably request screening of siblings and genetic counselling. All first degree relatives are screened in FPPH. It is thought that an individual in a family with FPPH has a 5–10% lifetime risk of developing PPH. Genetic testing is still not routine. DNA sequencing is necessary because mutations in the BMPR-2 gene appear to be “private” to each family.
Congenital heart disease
Correlating the physiological findings with structural observations in different types intracardiac abnormality has improved the accuracy with which immediate and long term outcome can be predicted with and without corrective surgery.19 However, prediction is still more difficult in young than in older children. Severe medial hypertrophy, although potentially reversible, increases the risk of postoperative pulmonary hypertensive crises. The most crucial factor in determining late outcome is the age at which repair is carried out. Most children operated upon by 9 months of age have a normal pulmonary vascular resistance one year after repair. After 2 years of age resistance may fall, but not to a normal level. These observations indicate vessel wall remodelling towards normality, continued growth, and a demonstrable improvement in endothelial function. Repairing an intracardiac abnormality in the presence of established disease accelerates the progression of disease and the onset of right ventricular failure and death. The effect of associated lesions, such as coarctation of the aorta, must also be taken into account, even when repaired earlier. If there is doubt about the likely outcome of surgical repair, then an open lung biopsy should clarify the position.
For patients with the Eisenmenger syndrome treatment is still largely empirical and includes:
long term oxygen treatment at home which gives subjective improvement and can increase survival
dipyridomole to reduce platelet aggregation, but it may also have a beneficial vasodilatory effect as a phosphodiesterase inhibitor
anticoagulation
venesection with plasma dilution in those with a high haematocrit is not used routinely but may afford symptomatic relief to some patients; frequent venesections causing iron deficiency can increase the risk of cerebrovascular accidents.
Treatment with prostacyclin is tempting, but its efficacy is not yet proven in patients with the classical Eisenmenger syndrome. Given intravenously this non-selective vasodilator can cause systemic hypotension in the presence of a pulmonary–systemic communication. Inhalation of a stable analogue would be more appropriate. Patients with advanced pulmonary vascular obstructive disease may, like those with PPH, be unable to increase their NO production on exercise. Chronic administration of L-arginine might be helpful if it could be shown conclusively that these patients have a relative substrate deficiency of NO production.
Calcium channel blockers are not used.
Scleroderma and other connective tissue disorders
Chronic intravenous epoprostenol treatment has improved exercise capacity and haemodynamics in adults,20 and children are treated in a similar fashion. Those with inflammatory disease are also given immunosuppressive drugs.
Finally, the only effective treatment for the very sick patient with pulmonary vascular disease of any aetiology who has failed medical treatment is lung transplantation. This is not usually an option in young children, but some older patients with PPH being treated as a bridge to transplantation have improved to such an extent that they are being treated medically long term rather than being transplanted. Since the results of transplantation are less than optimal, transplantation should only be considered when the expected survival time is less than the expected survival after transplantation. In future, we can hope that prompt early referral and effective treatment with the new and emerging therapies will postpone the need for transplantation indefinitely in many young people.
PERSPECTIVES ON FUTURE MANAGEMENT
There is an urgency to:
educate the medical professions to ensure earlier diagnosis and referral
improve non-invasive evaluation of disease severity, progression, and response to treatment in children
maximise the effect of current treatment, by selective, trialled combinations of drugs for use at different stages of disease
elucidate the role of endothelin receptor antagonists—the extent to which they can replace/be used with intravenous prostacyclin and the different analogues
develop new, stable prostacyclin analogues with a longer half life for oral and inhalational use and specific phosphodiesterase V inhibitors
explore novel treatments such as elastase inhibitors, gene therapy, and treatments based on exploitation of key signalling pathways identified by BMPR2 mutations in FPPH
stimulate growth of new, normal vessels, particularly in the young who have developed pulmonary hypertension before the lung fulfilled its growth potential and in whom growth potential is greater.
REFERENCES
Linked Articles
- Miscellanea