Article Text

Abstract
Background Pulmonary hypertension (PH) is frequently observed in patients with acute respiratory distress syndrome (ARDS) and it is associated with an increased risk of mortality. Both acid sphingomyelinase (aSMase) activity and interleukin 6 (IL-6) levels are increased in patients with sepsis and correlate with worst outcomes, but their role in pulmonary vascular dysfunction pathogenesis has not yet been elucidated. Therefore, the aim of this study was to determine the potential contribution of aSMase and IL-6 in the pulmonary vascular dysfunction induced by lipopolysaccharide (LPS).
Methods Rat or human pulmonary arteries (PAs) or their cultured smooth muscle cells (SMCs) were exposed to LPS, SMase or IL-6 in the absence or presence of a range of pharmacological inhibitors. The effects of aSMase inhibition in vivo with D609 on pulmonary arterial pressure and inflammation were assessed following intratracheal administration of LPS.
Results LPS increased ceramide and IL-6 production in rat pulmonary artery smooth muscle cells (PASMCs) and inhibited pulmonary vasoconstriction induced by phenylephrine or hypoxia (HPV), induced endothelial dysfunction and potentiated the contractile responses to serotonin. Exogenous SMase and IL-6 mimicked the effects of LPS on endothelial dysfunction, HPV failure and hyperresponsiveness to serotonin in PA; whereas blockade of aSMase or IL-6 prevented LPS-induced effects. Finally, administration of the aSMase inhibitor D609 limited the development of endotoxin-induced PH and ventilation-perfusion mismatch. The protective effects of D609 were validated in isolated human PAs.
Conclusions Our data indicate that aSMase and IL-6 are not simply biomarkers of poor outcomes but pathogenic mediators of pulmonary vascular dysfunction in ARDS secondary to Gram-negative infections.
- ARDS
- Bacterial Infection
- Cytokine Biology
- Innate Immunity
Statistics from Altmetric.com
Key messages
What is the key question?
Pulmonary hypertension and right ventricular dysfunction are prominent prognostic features of acute respiratory distress syndrome (ARDS) which, given the unclear pathophysiology and the lack of approved pharmacological therapies, demand the identification of new therapeutic targets.
What is the bottom line?
Blockade of either acid sphingomyelinase or IL-6 prevents the endothelial dysfunction, the failure of hypoxic pulmonary vasoconstriction and the hyperresponsiveness to serotonin induced by lipopolysaccharide (LPS) in vitro and, accordingly, the in vivo administration of the non-specific acid sphingomyelinase inhibitor D609 prevents the development of LPS-induced pulmonary hypertension.
Why read on?
This study identifies for the first time that acid sphingomyelinase and IL-6 are not simply biomarkers of outcome but rather pathogenic mediators of pulmonary vascular dysfunction and, as such, could represent novel therapeutic targets for ARDS secondary to Gram-negative infections.
Introduction
Sepsis is the most common risk factor for acute respiratory distress syndrome (ARDS).1 ,2 Acute lung injury (ALI)/ARDS is characterised by pulmonary oedema and alveolar collapse accompanied by hypoxic pulmonary vasoconstriction (HPV) failure, resulting in ventilation-perfusion mismatch and severe arterial hypoxaemia. Nearly 40 years ago, Zapol and Snider described for the first time3 that patients with ARDS frequently develop mild to moderate pulmonary hypertension (PH), which contrasts with the lowered systemic vascular resistance that characterises septic shock.4 ,5 Following this landmark study, pulmonary vascular dysfunction, clinically defined as an increase in transpulmonary gradient and augmented pulmonary vascular resistance, has gathered increasing attention. Earlier studies reported an incidence of PH and right ventricular dysfunction of up to 70%.6 In line with this, it has been shown that injurious mechanical ventilation is an important cause of pulmonary vascular injury in patients and in animal models of ALI/ARDS.7 However, in the setting of mechanical ventilation with lung protective strategies, pulmonary vascular dysfunction is still a prominent feature, with a prevalence of 20–25%, and is independently associated with poor outcomes in patients with ARDS.8 ,9 Taken together, these studies suggest that PH and right ventricular dysfunction can be caused not only by injurious mechanical ventilation but also by other mechanisms, including local alveolar hypoxia, inflammation and those associated with infection in ARDS.4 ,9
The mechanisms leading to acute PH in these patients are complex and most likely depend on the underlying cause of ARDS. However, endothelial dysfunction, hypoxia and imbalance of endogenous vasoactive factors, with increased production of pulmonary vasoconstrictors (endothelin 1, thromboxane A2 or serotonin) over vasodilators (nitric oxide (NO) or prostacyclin), are thought to play a major role.4 ,5 Given the association between infection and pulmonary vascular dysfunction in patients with ARDS, it is tempting to speculate that inflammation-mediated pulmonary vasoconstriction may also contribute to the increase in pulmonary artery pressure (PAP) in patients with ARDS.
Interleukin 6 (IL-6) and NO are two well recognised inflammatory mediators upregulated in sepsis and ARDS.10 ,11 Overproduction of NO by inducible NO synthase (iNOS) is believed to play a pivotal role in the reduced vascular contractility in endotoxin shock. However, PH develops despite iNOS induction in pulmonary arteries (PAs).12 Moreover, the inhibition of NOS activity seems to improve the general systemic haemodynamic situation but at the cost of raised pulmonary vascular resistance13 ,14 and increased mortality.15 On the other hand, IL-6 is one of the most relevant biomarkers in sepsis. Although IL-6 can activate both proinflammatory and anti-inflammatory pathways, IL-6 levels correlate with mortality risk from sepsis and ARDS.11 Interestingly, while IL-6 increases iNOS activity in aortic smooth muscle cells16 and it is associated with a drop in systemic blood pressure in patients with septic shock,17 the levels of IL-6 correlate with PAP in patients with PH secondary to COPD or cirrhosis18 ,19 and have a negative impact on survival in patients with pulmonary arterial hypertension (PAH).20 Indeed, a therapeutic potential for IL-6 blockade has been demonstrated by neutralising antibodies in experimental models of chronic PH.21 However, the exact role of IL-6 in the pathogenesis of pulmonary vascular dysfunction associated with sepsis has not yet been elucidated.
A growing body of evidence suggests an important role of sphingolipids in lung diseases.22 Thus, acid sphingomyelinase (aSMase), which is increased in critically ill patients,23 has been shown to contribute to the development of pulmonary oedema in experimental ALI.24–26 Notably, ceramide (the end product of SMase activity) has been shown to induce the production of IL-6, as effectively as IL-1β.27 In addition to its effects on the regulation of pulmonary inflammation and barrier function, ceramide is able to induce pulmonary vasoconstriction and seems to be a conserved effector mechanism in acute oxygen-sensing vascular tissues.28 ,29 Indeed, neutral SMase-derived ceramide production is an early and necessary event in the signalling cascade of acute HPV leading to vasoconstriction and increased PAP.28 However, whether aSMase contributes to the development of PH and HPV failure in ARDS is currently unknown.
We hypothesised that activation of aSMase and IL-6 production might contribute to the development of pulmonary vascular dysfunction induced by lipopolysaccharide (LPS). Therefore, in the present study we first used an in vitro approach to characterise the role of aSMase and IL-6 in the effects induced by LPS in isolated PA and cultured PA smooth muscle cells (PASMCs). Finally, the efficacy of an aSMase inhibitor, D609, was tested in vivo in a rat model of ALI induced by intratracheal instillation of LPS, used as a model of direct lung injury induced by Gram-negative bacteria.
Material and methods
An expanded Methods section is available in the online supplementary material.
Supplemental material
Vessel and cell isolation
PA were isolated from male Wistar rats or human lung tissue, as previously described.7 ,28 ,29 Freshly isolated PASMCs and primary cell cultures were obtained by enzymatic digestion, as described previously.28 ,29
PASMCs or whole pulmonary artery organ culture
Rat or human PA rings or rat PASMCs were pretreated with different pharmacological inhibitors or small interfering RNAs (siRNAs) before being exposed to LPS (1 μg/mL; Escherichia coli 055:B5), bacterial SMase (0.1 U/mL; Bacillus cereus) or IL-6 (30 ng/mL) for 24 hours, as detailed in the online supplementary material. 24 hours later, cell viability was evaluated by the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay30 and culture medium was frozen until further analysis.
Ceramide content detection by immunofluorescence
Ceramide content in freshly isolated PASMCs was quantified by immunocytochemistry, as previously described.29
Recording of vascular reactivity
PA rings were mounted in a wire myograph and exposed to hypoxia. Dose–response curves to serotonin, phenylephrine, endothelin 1 (ET-1) or acetylcholine were performed by cumulative addition, detailed in the online supplementary material.28 ,29
Animal model of LPS-induced lung injury
Adult male Wistar rats were treated with vehicle (DMSO) or D609 (50 mg/kg; intraperitoneally) for 30 min before intratracheal administration of LPS (3 mg/kg) or saline solution (n=6–12). Animals were randomly allocated to three experimental groups: (1) control group (DMSO-saline solution); (2) DMSO-LPS group and (3) D609-LPS group. 4 hours following instillation, rats were anaesthetised and PAP was registered in open-chest rats, as previously reported.28 Changes in oxygen saturation following partial airway occlusion by saline instillation was performed as detailed in the online supplementary material and described previously.31
Analysis of inflammatory markers
Levels of IL-6 and IL-1β were quantified by ELISA, NO production was estimated by the accumulation of nitrite, using the Griess reaction,32 ,33 and lung myeloperoxidase (MPO)26 and aSMase activity were measured as detailed in the online supplementary material.
Statistical analysis
Results are expressed as mean±SEM. Technical replicates were averaged to provide a single data point before any further analysis. Statistical analysis was performed using GraphPad Prism 5 as detailed in each figure legend. One-way ANOVA (for normally distributed data) followed by Bonferroni's post hoc test or non-parametric Kruskal–Wallis test followed by Dunn's multiple comparison test were used to compare three or more datasets. Repeated measures ANOVA was used to compare dose–response curves and one sample t-test to evaluate normalised datasets. A value of p<0.05 was considered statistically significant.
Results
LPS and exogenous SMase lead to ceramide production in freshly isolated PASMCs
Exogenous addition of SMase, which cleaves membrane sphingomyelin, produced a marked increase in ceramide content in freshly isolated rat PASMCs and served as a positive control (figure 1A, B). Furthermore, incubation with LPS for 30 min increased ceramide content by 40%.

Lipopolysaccharide (LPS) and exogenous addition of bacterial sphingomyelinase (SMase) induce an increase in ceramide and interleukin 6 (IL-6) production in rat pulmonary artery smooth muscle cells (PASMCs). (A) Representative pictures and (B) quantification of ceramide content (arbitrary units) in freshly isolated rat PASMC incubated under control conditions (n=3 coverslips with at least 10 cells, each derived from a different animal) or exposed to LPS (1 μg/mL; n=3) or SMase (0.1 U/mL; n=4) for 30 min and immunostained with a monoclonal ceramide-specific antibody (15B4). Data are shown as the mean±SEM of the averaged FITC-fluorescence intensity relative to cell surface. (C and D) IL-6 levels (ng/mL) released in the culture media by cultured rat PASMCs incubated under control conditions or exposed to increasing concentrations of LPS (C) or exogenous SMase (D) for 24 hours. Data are shown as the mean±SEM from n=3 experimental runs performed in triplicate. *p<0.05, **p<0.01 and ***p<0.001 versus control (analysed with one-way ANOVA followed by the Dunnett's post hoc test).
LPS stimulates IL-6 production in PASMCs via an aSMase-dependent and TAK1-dependent pathway
LPS and exogenous SMase induced a concentration-dependent increase in IL-6 production in cultured rat PASMCs (figure 1C, D). Once established that LPS increases ceramide content and that exogenous SMase can mimic the inflammatory response induced by LPS in PASMCs, we pharmacologically dissected the signalling pathways mediating these effects. As expected, the selective toll-like receptor 4 (TLR4) antagonist TAK-242 fully blocked the LPS-induced IL-6 release but had no effect on that induced by SMase (figure 2A). IL-6 production stimulated by either LPS or SMase showed a similar sensitivity to dexamethasone (figure 2B) and were unaffected by the iNOS inhibitor 1400W (figure 2C). Since transforming growth factor β activated kinase (TAK1) is activated by LPS34 or ceramide,35 we assessed the role of this kinase in the production of IL-6. As shown in figure 2D, the TAK1 inhibitor 5Z-7-oxozeaenol exerted a dose-dependent inhibitory effect in either LPS or SMase treated cells. Notably, IL-6 induced by either LPS or SMase was inhibited by two chemically unrelated aSMase inhibitors, D609 and desipramine (figure 2E, F). Since both drugs may interact with targets other than aSMase,36 we confirmed the role of aSMase by two different siRNA targeting SMPD1, the gene encoding aSMase. As shown in figure 2G–H, both siRNAs reduced SMPD1 mRNA expression by 75–80% and significantly inhibited the release of IL-6 induced by LPS. As expected, downregulation of endogenous aSMase had no effect on the responses to exogenous SMase.

Role of acid sphingomyelinase (aSMase) in lipopolysaccharide (LPS)-induced IL-6 release by rat pulmonary artery smooth muscle cells (PASMCs). Rat PASMCs were incubated under control conditions or exposed to LPS (1 μg/mL; black bars) or exogenous SMase (0.1 U/mL; grey bars) for 24 hours before quantifying interleukin 6 (IL-6) levels (ng/mL) released in the culture media. (A–F) Effects of different pharmacological inhibitors on the secretion of IL-6 stimulated by LPS (black bars) or bacterial SMase (grey bars). Rat PASMCs were treated with increasing concentrations of: the toll-like receptor 4 (TLR4) receptor antagonist TAK-242 (A; n=5 experimental runs), the glucocorticoid dexamethasone (B; n=3–5), the inducible nitric oxide synthase (iNOS) inhibitor 1400W (C; n=4), the transforming growth factor β activated kinase (TAK1) inhibitor 5Z-7-oxozeaenol (D; n=3–5) or the aSMase inhibitors D609 (E; n=3) and desipramine (F; n=3–4). (G and H) Effects of two different siRNAs targeting rat SMPD1 on SMPD1 mRNA levels (G) and on the release of IL-6 induced by LPS and exogenous SMase (H) from three different experimental runs. Expression of SMPD1 was analysed by RT–PCR and results were normalised to GAPDH and expressed as a percent of mean values of PASMCs treated with random siRNA. Production of IL-6 was expressed as a percentage of the response induced by LPS or SMase (control/random), *p<0.05, **p<0.01 and ***p<0.001 versus LPS or SMase control/random (one sample t-test).
Induction of iNOS activity in rat PA by LPS is independent of aSMase, TAK1 and IL-6
LPS induced a robust increase in the levels of nitrite (figure 3A) which was inhibited by 1400W (figure 3B). In contrast, exogenous SMase was unable to exert any significant effect on iNOS activity (figure 3A). Furthermore, the increase in iNOS activity induced by LPS was resistant to 5Z-7-oxozeaenol (figure 3B), suggesting that the induction of iNOS is not mediated via activation of TAK1. However, the two aSMase inhibitors produced disparate results; while desipramine had no effect on iNOS activity, D609 partially reduced the effects induced by LPS (figure 3B).

Lipopolysaccharide (LPS), but not exogenous sphingomyelinase (SMase), promotes inducible nitric oxide synthase (iNOS) activity in whole pulmonary arteries (PAs). (A) Rat PA rings were incubated in the absence (control; n=17) or in the presence of LPS (1 µg/mL; n=20) or exogenous SMase (0.1 U/mL; n=7) for 48 hours and iNOS activity was assessed by measurement of nitrite (breakdown product of NO) by the Griess assay. (B) Rat PA rings were pretreated for 30–45 min prior to addition of LPS with 1400W (10−4 M; n=3), 5Z-7-oxozeaenol (10−7 M; n=4), D609 (10−4 M; n=8) and desipramine (DESI; 10−5 M; n=5). Data were expressed as a percentage of the response induced by LPS (control). *p<0.05 and ***p<0.001 versus control (one-way ANOVA followed by Dunnett's post hoc test or one sample t test). (C) Contractile responses to phenylephrine in isolated PA incubated 24 hours in the absence (control; n=35) or presence of LPS (1 µg/mL; n=25). Effects of (D) the iNOS inhibitor 1400W (10−4 M; n=9–10), (E) bacterial SMase (0.001 U/mL; n=18), (F) D609 (10−4 M; n=5–6), (G) recombinant IL-6 (30 ng/mL; n=20–21), or (H) the neutralising anti-IL-6 antibody (0.56 µg/mL; n=15–18). Data are shown as active effective pressure (mN/mm2). *p<0.05, **p<0.01 and ***p<0.001 versus control (repeated measures ANOVA followed by Bonferroni’s post hoc test).
One of the main known functional consequences of vascular iNOS induction is a reduced contraction to α-adrenoceptor stimulation, a feature shared by pulmonary and systemic arteries.10 Likewise, LPS markedly reduced the pulmonary vasoconstriction to phenylephrine (figure 3C) and this effect was fully prevented by the selective iNOS inhibitor (figure 3D). Notably, vasoconstriction induced by phenylephrine was not altered following treatment with SMase or recombinant IL-6 (figure 3E, G). In line with this, neither D609 nor the IL-6 neutralising antibody prevented LPS-induced hyporesponsiveness to phenylefrine (figure 3F, H). A goat IgG, an isotype control antibody, did not affect LPS-induced effects discarding non-specific effects (see online supplementary figure S1). The effects of desipramine on vascular reactivity were not analysed because of its known properties as a Ca2+channel blocker.37
LPS enhances serotonin-induced pulmonary vasoconstriction via aSMase and IL-6
Serotonin and ET-1 are two vasoactive factors implicated in the pathogenesis of PAH and upregulated in sepsis and ARDS.4 ,5 ,38 In contrast to the effects observed on adrenergic-induced pulmonary vasoconstriction, treatment with LPS preserved the contractile responses to ET-1 (see online supplementary figure S2) and markedly increased pulmonary vasoconstriction induced by serotonin (figure 4A, C). Exogenous SMase and IL-6 enhanced the contractile responses to serotonin (figure 4E, G), mimicking the effects induced by LPS. Furthermore, hyperresponsiveness to serotonin was insensitive to iNOS inhibition (figure 4D) but was prevented by D609 or the IL-6 neutralising antibody (figure 4B, F, H). It should be noted that D609 prevented the hyperresponsiveness and unmasked a hyporesponsiveness to serotonin in LPS-treated arteries.

Acid sphingomyelinase (aSMase) and interleukin 6 (IL-6) mediate the hyperresponsiveness to serotonin induced by lipopolysaccharide (LPS) in rat pulmonary arteries (PAs). Representative tracings showing the contractile responses of PAs incubated in the absence or presence of LPS (A) or D609 (B) and mounted in a wire myograph. (C) Mean contractile responses to serotonin in isolated PAs incubated for 24 hours in the absence (control; n=35) or presence of LPS (1 µg/mL; n=25) from n=31–40. Effects of (D) 1400W (10−4 M; n=8–9), (E) bacterial SMase (0.001 U/mL; n=14), (F) D609 (10−4 M; n=5–6), (G) recombinant IL-6 (30 ng/mL; n=20–21) and (H) the neutralising anti-IL-6 antibody (0.56 µg/mL; n=15–20). Data are shown as active effective pressure (mN/mm2). *, ** and ***p<0.05, p<0.01 and p<0.001 versus control (repeated measures ANOVA followed by Bonferroni’s post hoc test).
iNOS and aSMase/IL-6 pathways modulate the failure of HPV and endothelial dysfunction induced by LPS
As opposed to systemic vascular beds, hypoxia induces vasoconstriction in the pulmonary vasculature. As we have previously shown,28 in the absence of other vasoconstrictor agents, acute exposure to hypoxia induced a contractile response (ie, HPV) in isolated PAs incubated under controlled conditions (figure 5A, B). In line with previous reports,39 ,40 incubation with LPS fully blocked HPV (figure 5A, B). Exogenous SMase and IL-6 mimicked the effects induced by LPS on HPV (figure 5C). Treatment with the iNOS inhibitor 1400W, the aSMase inhibitor D609 or the IL-6 neutralising antibody prevented the impairment of HPV by LPS (figure 5B). Moreover, the effects induced by D609 were concentration dependent and the highest concentration tested (10−4 M) preserved and significantly potentiated HPV in LPS-treated PAs (figure 5B and see online supplementary figure S3A). To translate our findings into human vessels, additional experiments were performed using human PAs. Data shown in figure 5D confirmed that incubation with LPS inhibited HPV, as seen in rat PA, and again treatment with D609 preserved and significantly potentiated this response in human PAs.

Inhibition of inducible nitric oxide synthase (iNOS), acid sphingomyelinase (aSMase) and interleukin 6 (IL-6) prevents the impairment of hypoxic pulmonary vasoconstriction (HPV) induced by lipopolysaccharide (LPS) in vitro. (A) Representative tracings showing the time course and reversibility of the contractile responses of pulmonary arteries (PAs) exposed to hypoxia at time 0 and incubated under control conditions or in the presence of LPS (left) or D609 (right). (B) Mean contractile responses to hypoxia in rat PAs incubated under control conditions (white bars; n=35) or in the presence of LPS (1 µg/ml; black bars; n=25) following treatment with the iNOS inhibitor 1400W (10−4 M; n=5–9), the neutralising anti-IL-6 antibody (0.56 µg/mL; n=15–18) or D609 (10−4 M; n=17–18). (C) Effects of bacterial sphingomyelinase (SMase 0.001 U/mL; n=17) and recombinant IL-6 (30 ng/mL; n=20) on HPV in rat PAs. (D) Mean HPV in isolated human PA (n=4–13 different patients) incubated in the absence (control) or the presence of LPS (1 µg/mL) or pretreated with D609 (10−4 M) 45 min prior to the addition of the agonist. Data are shown as active effective pressure (mN/mm2). *, ** and ***p<0.05, p<0.01 and p<0.001 versus control and †† and ††† p<0.01 and p<0.001 versus LPS (analysed by one-way ANOVA or two-way ANOVA, as appropriate, followed by Bonferroni’s post hoc test).
Endothelial dysfunction is a common feature in systemic and PAs exposed to endotoxin.4 ,10 Accordingly, the relaxant responses to acetylcholine were blunted in LPS-treated PAs (figure 6A) and this effect was fully prevented by 1400W (figure 6B). LPS-induced endothelial dysfunction was also prevented by treatment with D609 whereas the IL-6 neutralising antibody exhibited a modest protective effect (figure 6C, D). In line with this, exposure to SMase and IL-6 also induced endothelial dysfunction (figure 6E). Finally, treatment with D609 also preserved endothelial function in human PAs (figure 6F).

Pulmonary endothelial dysfunction induced by lipopolysaccharide (LPS) in vitro is prevented by inhibition of inducible nitric oxide synthase (iNOS), acid sphingomyelinase (aSMase) and interleukin 6 (IL-6). Concentration-dependent relaxation induced by the endothelium-dependent vasodilator acetylcholine in control (n=35) or LPS-treated (n=25) rat pulmonary artery (PA) rings incubated in the absence from n=31–40 (A) or the presence of (B) the iNOS inhibitor 1400W (10−4 M; n=9–10), (C) D609 (10−4 M; n=11–17) or (D) the neutralising anti-IL-6 antibody (0.56 µg/mL; n=15–20). (E) Relaxation induced by acetylcholine in the absence (control; n=19) or in the presence of bacterial sphingomyelinase (SMase 0.001 U/mL; n=18) or recombinant IL-6 (30 ng/mL; n=17). (F) Effects of pretreatment with D609 (10−4 M) on the relaxation induced by acetylcholine in human PAs (n=4–13 different patients) incubated under control conditions or exposed to LPS. Results are expressed as a percentage of the relaxation induced by acetylcholine. *p<0.05, **p<0.01 and ***p<0.001 versus control and †p<0.05 and †††p<0.001 versus LPS (repeated measures ANOVA followed by Bonferroni’s post hoc test).
D609 inhibits LPS-induced PH in vivo
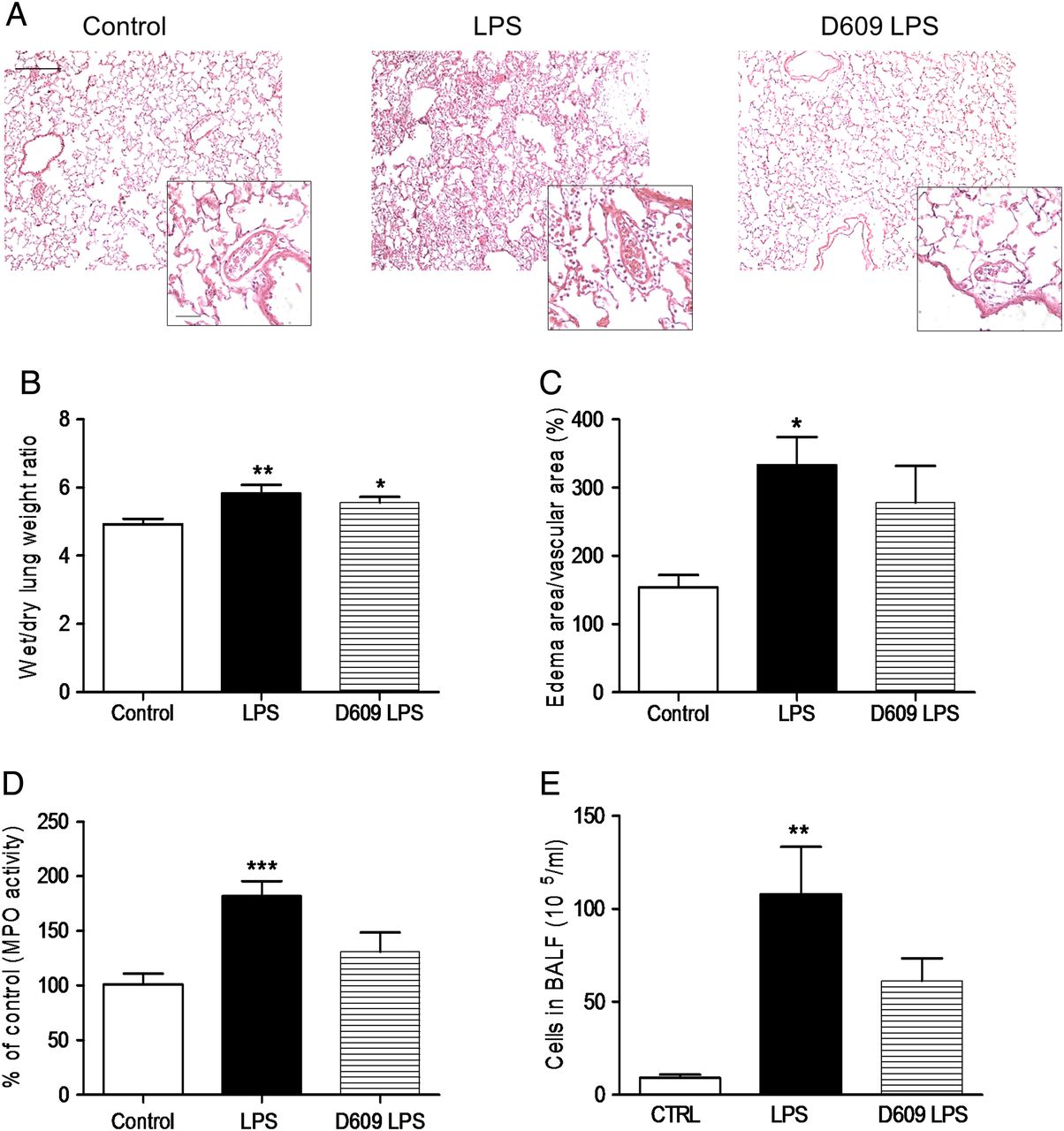
Adult healthy male Wistar rats were treated with vehicle (DMSO) or D609 (50 mg/kg body weight; intraperitoneally) for 30 min before intratracheal administration of LPS (3 mg/kg body weight) or saline solution (control). All the animals survived to the procedures. Intratracheal instillation of LPS increased pulmonary vascular permeability, as evidenced by the formation of perivascular oedema and lung weight gain. Notably, while lung oedema was relatively insensitive to D609, markers of lung inflammation (such as MPO activity, number of cells in bronchoalveolar lavage fluid (BALF) or production of IL-1β) were significantly attenuated by treatment with D609 (see figure 7 and online supplementary figure S4).

Effects of treatment with D609 on markers of lung oedema and pulmonary inflammation in an in vivo model of lipopolysaccharide (LPS)-induced acute lung injury. (A) Representative photomicrographs of lung sections stained with H&E (×10; scale bar: 250 μm) and insets showing pulmonary arteries (×40; scale bar: 50 μm) from control, LPS and D609 plus LPS-treated rats. Effects of intraperitoneal administration of D609 (50 mg/kg) on wet to dry lung weight ratio (B), size of perivascular oedema (measured as % of oedema area/total vascular area) (C), whole lung myeloperoxidase activity (MPO; D) and number of cells in bronchoalveolar lavage fluid (BALF) (E) following intratracheal administration of LPS (3 mg/kg). Results are shown as the mean±SEM (n=3–9). Data were analysed with the non-parametric Kruskal–Wallis test followed by Dunn’s test (B, C and E). *p<0.05, **p<0.01 and ***p<0.001 versus control.
In line with our in vitro assays, LPS increased aSMase activity, but not protein expression levels (data not shown), in lung homogenates from control but not from D609-treated rats (figure 8A). Similarly, LPS also induced an increase in the amount of IL-6 in whole lung homogenates which was significantly reduced by D609 (figure 8B). Despite direct pulmonary delivery, LPS also induced a modest but significant increase in plasma IL-6 which was prevented by D609 (see online supplementary figure S4). Moreover, intratracheal instillation of LPS resulted in an increase in PAP which was prevented by treatment with D609 (figure 8C, D). The strong correlation found between mean PAP and pulmonary, but not systemic, IL-6 levels, suggests that lung-derived IL-6 is an important mediator of LPS-induced PH (figure 8E). Similar results were found with IL-1β and ET-1, a well known mediator of endotoxin-induced PH.4 ,5 Thus, lung IL-1β and ET-1 levels were prevented by treatment with D609 and correlated with mean PAP, whereas no significant associations were found between systemic levels and haemodynamic alterations (see online supplementary figure S4). Taken together, these data suggest that the beneficial effects of D609 on pulmonary pressure are due to direct effects on the lung.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
D609 inhibits lung acid sphingomyelinase (aSMase) activity, interleukin 6 (IL-6) production and limits lipopolysaccharide (LPS)-induced pulmonary hypertension in vivo. Effects of intraperitoneal administration of D609 (50 mg/kg) on (A) whole lung aSMase activity and (B) IL-6 production following intratracheal administration of LPS (3 mg/kg). Data were collected from n=5–8 animals. (C) Representative recordings of pulmonary artery pressure (PAP) in control, LPS and D609 plus LPS treated rats (n=7, 9 and 5, respectively). (D) Averaged values of diastolic, systolic and mean PAP registered in vivo 4 hours after instilling intratracheally saline solution (control) or LPS (3 mg/kg). (E) Correlation between pulmonary IL-6 levels and mean PAP in control, LPS and D609 plus LPS treated rats (n=4–5). (F) Time course of oxygen saturation (% of O2) following partial airway occlusion by intratracheal instillation of saline solution. Data are shown as the mean±SEM. *p<0.05, **p<0.01 and ***p<0.001 versus control and †p<0.05 and ††p<0.01 versus LPS analysed with repeated measures ANOVA (F) or one-way ANOVA followed by the Bonferroni's post hoc test for normally distributed data (B and D) or non-parametric Kruskal–Wallis test followed by Dunn’s multiple comparison test (A).
Finally, to test whether the impairment of HPV observed in vitro translated into ventilation-perfusion mismatching in vivo, we used a model of partial airway occlusion. As shown in figure 8F, intratracheal administration of 100 µL of saline solution induced a rapid but transient hypoxaemia. The maximum decline in oxygen saturation levels was similar among all the groups. By contrast, the recovery, which reflects blood redistribution away from the unventilated/fluid filled alveoli (ie, HPV) was significantly delayed in rats challenged with LPS but not in D609-treated rats.
Discussion
aSMase and IL-6 levels are frequently elevated in critically ill patients and correlate with worst outcomes.11 ,23 Our results demonstrate that LPS stimulates ceramide and IL-6 production in PASMCs. These effects are independent of iNOS and are associated with the induction of pulmonary vascular dysfunction in vitro and PH in vivo. Exposure to exogenous SMase and recombinant IL-6 in vitro mimics the effects induced by LPS whereas their blockade prevents these effects. Furthermore, the non-specific aSMase inhibitor D609 prevents the development of LPS-induced PH and HPV failure in vivo. Importantly, the protective effects of D609 were validated in isolated human PA. Our data provide evidence for the role of aSMase and IL-6 not only as biomarkers of poor outcomes but also as pathogenic mediators of pulmonary vascular dysfunction in ARDS secondary to Gram-negative infections.
The recently described association between pulmonary vascular dysfunction and mortality in patients with ARDS highlights the utmost need to understand the pathophysiology of PH in ARDS.4 ,5 ,8 ,9 Despite the use of protective lung ventilation, the development of PH and acute cor pulmonale is still prevalent in patients with ARDS and seems to be associated with infection.9 Therefore, we have evaluated the effects of the inflammatory stimuli LPS on rat and human PAs. Our results confirm that LPS inhibited pulmonary vasoconstriction induced by phenylephrine or hypoxia, which reflects an excess vasodilation due to iNOS-derived NO. However, the endothelium-dependent and NO-dependent relaxation to acetylcholine was blunted, the vasoconstriction induced by ET-1 was preserved and serotonin-induced vasoconstriction was significantly increased. Why LPS increased PAP in vivo (figure 8) despite inducing hyporesponsiveness to phenylephrine in vitro is striking. However, given the relatively weak active force elicited by phenylephrine, compared with serotonin or ET-1, it is tempting to speculate that induction of endothelial dysfunction in the setting of normal or enhanced contractile responses might contribute to the increase in PAP seen after LPS administration in vivo. These results contrast with those obtained in systemic arteries, where LPS induces the expression of iNOS and causes a general impairment in vascular contractility, which ultimately explains the refractory drop in blood pressure induced by sepsis.10 ,13 However, our data are in agreement with findings in other models of septic shock or ALI, where the pulmonary circulation exhibits different degrees of vasoconstriction depending on the vasoactive factor analysed.7 ,12 ,41
The role of iNOS in septic PH remains unclear. Thus, whereas iNOS induction might protect against PH by counteracting the action of vasopressor agents, it seems to contribute to the development of endothelial dysfunction and the impairment of HPV and ventilation-perfusion mismatching.10 ,12 ,39 ,40 The effects observed with the iNOS inhibitor 1400W are in line with numerous studies showing that the induction of iNOS inhibits α-adrenergic-mediated vasoconstriction and endothelial function, probably due to the formation of peroxynitrite, in pulmonary and systemic arteries.7 However, the role of iNOS in HPV is more controversial. The partial protection found in this and previous studies39 ,40 suggests that the HPV impairment during endotoxemia must be mediated, at least in part, by other mechanisms other than simple pulmonary vasodilation as a result of excess NO production. However, the fact that the hyperresponsiveness to serotonin persisted despite iNOS inhibition supports the notion that serotonin-induced contraction is rather resistant to the induction of iNOS in PAs.7
Among the wide range of inflammatory mediators involved in ARDS, we focused on IL-6 as a well recognised biomarker for poor outcomes in sepsis and ARDS.11 Furthermore, IL-6 appears to be increased in patients with different forms of PH,18–20 it promotes PASMC proliferation and its blockade limits vascular remodelling and PH in experimental models.21 ,42 ,43 Our study corroborates the ability of endotoxin to induce IL-6 production in PASMCs through a mechanism which is sensitive to glucocorticoids and aSMase inhibitors but is partially resistant to iNOS inhibition. An important finding of our study is that, in addition to the previously reported effects,42 ,43 IL-6 is able to directly modulate pulmonary vascular tone. Thus, incubation with IL-6 (30 ng/mL; concentration within the range produced by PASMCs upon LPS challenge) increased the contractile responses to serotonin, inhibited HPV and reduced the relaxant responses to acetylcholine in isolated PAs, thereby mimicking the effects of LPS. Induction of endothelial dysfunction by IL-6 has been shown in other vascular beds,44 ,45 through a mechanism involving inhibition of Akt and stabilisation of interactions between endothelial NOS and caveolin 1.46 ,47 However, in contrast to the effects observed in systemic arteries,48 IL-6 did not reduce the contractile responses to phenylephrine but potentiated those induced by serotonin in isolated rat PAs. Furthermore, to the best of our knowledge, this is the first study confirming a direct role of IL-6 in regulating HPV. Altogether, these findings suggest that increased levels of IL-6 might directly contribute to the development of pulmonary vascular dysfunction and ventilation-perfusion mismatch by impairing HPV. Accordingly, treatment with an anti-IL-6 neutralising antibody fully prevented the hyperresponsiveness to serotonin and partially prevented the endothelial dysfunction and HPV failure following LPS challenge.
aSMase and ceramide have recently emerged as potential mediators of inflammation and increased endothelial vascular permeability in ALI.24–26 In line with this, we observed that LPS rapidly increased ceramide content in PASMCs and inhibition of aSMase by either pharmacological inhibitors or siRNA prevented the increase in IL-6 induced by LPS. In addition, the exposure to exogenous SMase reproduced many of the effects induced by LPS, including IL-6 production, hyperresponsiveness to serotonin, endothelial dysfunction and HPV inhibition. Furthermore, inhibition of aSMase with D609 prevented these alterations in PA exposed to LPS. Interestingly, exogenous SMase failed to increase iNOS activity as evidenced by the lack of effects on nitrite levels and on phenylephrine-induced contraction. These findings are in agreement with previous reports showing that ceramide by itself was unable to induce iNOS activity but rather acted as a potentiating mechanism for other inflammatory stimuli.32 To test this hypothesis, we examined the contribution of aSMase to the effects induced by LPS on iNOS activity. Consistent with previous studies,33 D609 but not desipramine reduced LPS-induced iNOS activity (figure 3B). Desipramine inhibits aSMase by stimulating its proteolytic degradation whereas D609 blocks its activation by inhibiting phosphatidylcholine (PC)-specific phospholipase C (PC-PLC).36 Therefore, our data suggest that PC-PLC activation, but not aSMase activation, is essential for the induction of iNOS by LPS in PASMCs.
HPV is frequently impaired in patients with ARDS, resulting in ventilation-perfusion mismatch leading to severe arterial hypoxaemia. However, the vasoconstrictor response to hypoxia shows a high variability among patients and experimental models, with studies reporting decreased39 ,40 and increased7 ,12 HPV. Our results are in agreement with former studies since we have confirmed the ability of LPS to inhibit HPV in vitro in human and rat PAs. Furthermore, the partial protective effects of 1400W (present results and)39 ,40 and IL-6 neutralising antibody (present study) suggest that, in addition to iNOS, IL-6 is involved in the failure of HPV induced by LPS. In line with this, we report for the first time that D609 prevented and significantly potentiated the vasoconstrictor responses to hypoxia in human and rat PAs. Although the mechanism underlying this effect remains unclear, the identification of aSMase as a novel mediator for the failure of HPV in rat and human PAs may have an added relevance in human disease since induction of iNOS expression and NO production in response to endotoxin is lower in human cells than in rodents.10
Altogether, our in vitro data suggested that inhibition of the aSMase/IL-6 pathway could be an effective approach to limit endotoxin-induced PH. To test this hypothesis we analysed the effects of D609 in vivo in a model of direct lung injury induced by intratracheal instillation of LPS. As expected, administration of D609 inhibited lung aSMase activity and reduced the pulmonary levels of IL-6, corroborating some of our findings in vitro. Notably, D609 also preserved HPV and limited the increase in PAP in this in vivo model. In addition to the effects observed in the lungs, intratracheal instillation of LPS induced a systemic inflammatory response. However, the fold changes induced by LPS in pulmonary IL-6 and IL-1β (100-fold and 20-fold, respectively) were found to exceed those in the peripheral circulation (1.5-fold and 3-fold, respectively). This gradient suggests that the production of these cytokines is mainly confined within the lungs and that the amount of LPS (or interleukins) spill over into the systemic circulation is minor. Furthermore, the lack of correlation between circulating levels of either IL-6 or IL-1β and mean PAP suggests that the beneficial effects of D609 on pulmonary pressure are due to direct effects on the lung. It should be noted, however, that LPS increases the ET-1 levels by the same order of magnitude in lung tissue and plasma, and that the increase in plasma is resistant to aSMase inhibition. The origin and the effect of this circulating ET-1 is still unclear, however a recent study has demonstrated that the increase in serum ET-1 levels induced by LPS is regulated by the type I interferon receptor IFNAR1,38 a pathway which may be independent of aSMase.
Several studies have previously focused on the anti-inflammatory effects of D609 and its ability to limit the formation of pulmonary oedema in experimental models of ALI.24 ,25 Indeed, we found that treatment with D609 significantly reduced some markers of lung injury (including lung MPO activity, number of cells in BALF and levels of IL-6 and IL-1β) but not others (only a trend for a reduction in lung water content and perivascular oedema was observed). This lack of significant protective effects on pulmonary oedema contrasts with previous reports describing the ability of D609 to prevent the formation of lung oedema induced by platelet-activating factor, endotoxin or acid instillation.24 However, it should be noted that in the study by Göggel et al,24 multiple doses of D609 (four doses every 30 min) only exerted a partial protective effect and that complete protection against lung oedema was only achieved when both aSMase and cyclooxygenase pathways were inhibited. Therefore, the lack of protective effects against lung oedema in our animal model could be explained by the different dosing regimen between both studies. Moreover, our study identifies a novel protective effect of D609 on pulmonary vascular function. Anti-IL-6 therapies have demonstrated therapeutic efficacy in several inflammatory diseases but their use in infectious disease is controversial due to the potential impairment of immune responses. However, recent reports suggest a dose dependency of anti-IL-6 for a beneficial outcome.49 In this regard, the dose of D609 used in this study only reduced IL-6 levels by 50%, but this reduction was effective for limiting PH. Whether higher doses of D609 or other therapeutics targeting aSMase or IL-6 would have an added benefit in ALI while allowing an adequate amount of IL-6 for maintaining host–defence responses remains to be determined.
There are several limitations in this study. First, we did not measure the systemic blood pressure and we did not perform an echocardiographic study. Therefore, we cannot dismiss the fact that the protective effects of D609 on the pulmonary circulation could be explained, at least in part, by a general improvement in systemic haemodynamics or right ventricular function. Second, we have demonstrated that the effects of D609 in vitro are mediated by inhibition of aSMase. However, the effects induced by D609 can be related to several mechanisms of actions, including inhibition of PC-PLC, aSMase, sphingomyelin synthase or antioxidant effects. Therefore, we cannot rule out that other mechanisms different from inhibition of aSMase could contribute to the protective effects of D609 observed in vivo. Finally, ARDS can have either a pulmonary (ie, pneumonia or gastric aspiration) or extrapulmonary (sepsis or haemorrhagic shock) origin. Thus, the pathophysiology and severity of the disease may be influenced by its aetiology. Therefore, further research is needed to elucidate whether aSMase and IL-6 have a role in the pathogenesis of pulmonary and extrapulmonary ARDS.
In summary, the present study identifies two divergent signalling pathways after TLR4 activation, which are involved in endotoxin-induced pulmonary vascular dysfunction: (1) the canonical iNOS induction pathway, responsible for depressed responses to α-adrenoceptor activation and affecting HPV and endothelial dysfunction; and (2) a novel aSMase-TAK1-IL-6 pathway, playing a role in serotonin-induced hyperresponsiveness, reduced HPV and endothelial dysfunction. The latter seems to play an important role in endotoxin-induced increase in PAP. Thus, drugs targeting aSMase and/or IL-6 might represent new strategies to limit PH and improve gas exchange by pharmacological enhancement of HPV in lung injury associated with bacterial sepsis.
References
Footnotes
Contributors Involved in the conception, hypothesis delineation, and design of the study: RP, AC, FP-V, LM; performed the experiments or analysed the data: RP, BB, EM, VL-A, DM-C, AM-R, BdON, RH, JÁL, LM; interpretation of the data: RP, BB, EM, RH, JÁL, AC, FP-V and LM; wrote the paper or made substantial contribution prior to submission: RP, AC, FP-V and LM.
Funding This work was supported by the Instituto de Salud Carlos III (PI15/01100 and Miguel Servet Program: CP12/03304 to LM and predoctoral CIBERES grant to RP), the Spanish Ministry of Economy and Competitiveness (predoctoral FPI grant BES-2012–051904 to DM-C; research grants SAF2011-28150 to FP-V and SAF 2014-55399 to FP-V and AC) and co-financed by ERDF (FEDER) Funds from the European Commission, ‘A way of making Europe’, and Marie Curie European Reintegration Grant within the 7th European Community Framework Programme (PERG05-GA-2009–249165 to FP-V and LM).
Competing interests None declared.
Patient consent Obtained.
Ethics approval Ethical approvals were obtained from the human research ethics committee of the Hospital Universitario de Getafe (human samples) and experimental research ethics committee from the Universidad Complutense de Madrid.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement We have additional data regarding aSMase protein expression which are available upon request.