Article Text

Abstract
Background Serum KL6/mucin 1 (MUC1) has been identified as a potential biomarker in idiopathic pulmonary fibrosis (IPF), but the role of MUC1 intracellular bioactivation in IPF is unknown.
Objective To characterise MUC1 intracellular bioactivation in IPF.
Methods and results The expression and phosphorylation of Thr41 and Tyr46 on the intracellular MUC1-cytoplasmic tail (CT) was increased in patients with IPF (n=22) compared with healthy subjects (n=21) and localised to fibroblasts and hyperplastic alveolar type II cells. Transforming growth factor (TGF)-β1 phosphorylated SMAD3 and thereby increased the phosphorylation of MUC1-CT Thr41 and Tyr46 in lung fibroblasts and alveolar type II cells, activating β-catenin to form a phospho-Smad3/MUC1-CT and MUC1-CT/β-catenin nuclear complex. This nuclear complex promoted alveolar epithelial type II and fibroblast to myofibroblast transitions, as well as cell senescence and fibroblast proliferation. The inhibition of MUC1-CT nuclear translocation using the inhibitor, GO-201 or silencing MUC1 by siRNA, reduced myofibroblast transition, senescence and proliferation in vitro. Bleomycin-induced lung fibrosis was reduced in mice treated with GO-201 and in MUC1-knockout mice. The profibrotic lectin, galectin-3, directly activated MUC1-CT and served as a bridge between the TGF-β receptor and the MUC1-C domain, indicating TGF-β1-dependent and TGF-β1-independent intracellular bioactivation of MUC1.
Conclusions MUC1 intracellular bioactivation is enhanced in IPF and promotes fibrotic processes that could represent potential druggable targets for IPF.
- idiopathic pulmonary fibrosis
- interstitial fibrosis
Statistics from Altmetric.com
Key messages
What is the key question?
What is the role of mucin 1 (MUC1) intracellular bioactivation in promoting lung fibrosis in idiopathic pulmonary fibrosis (IPF)?
What is the bottom line?
The MUC1 intracellular cytoplasmic tail is activated in experimental and human pulmonary fibrosis and leads to altered alveolar type II epithelial cell and fibroblast function in IPF.
Why read on?
This is the first report that identifies MUC1 intracellular bioactivation as a potential drug target in IPF.
Background
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive and fatal form of fibrotic interstitial lung disease of unknown aetiology that progresses until death at 2.5–5 years after diagnosis.1 IPF is characterised by heterogeneous lung lesions at various stages of the disease course, with foci of proliferative fibroblasts, myofibroblast formation, abnormal lung epithelial cells and overwhelming matrix accumulation in the lung interstitium.2 The origins of the invasive lung myofibroblasts, and the activation thereof, are not well known, but probably include activation of lung-resident fibroblasts and mesenchymal/myofibroblast transformation of alveolar type II (ATII) epithelial cells.3 Senescence of alveolar epithelial cells and fibroblasts appears to be a central phenotype that promotes lung fibrosis through the increased production of a broad range of growth factors, cytokines, chemokines and matrix metalloproteinases as well as acquired apoptosis resistance.4
A growing number of studies have been performed to identify protein-based and cell-based predictors of IPF disease.5 Elevated serum levels of several proteins have been associated with poorer prognosis in IPF, including surfactant proteins A (SP-A) and D (SP-D), and mucin 1 (MUC1).6 The transmembrane mucin, MUC1, is a large glycoprotein that acts as a membrane receptor consisting of three domains: (1) an extracellular domain, (2) a single transmembrane region and (3) a cytoplasmic tail (CT). The extracellular region (MUC1-N or α-chain) contains the KL-6 epitope domain, which is cleaved from the cell surface in response to injury and released into the surrounding environment.7 KL-6 is increased in the serum, bronchoalveolar lavage fluid (BALF) and lung tissue of patients with IPF.7 Isolated KL-6 can promote lung fibroblast migration, proliferation and myofibroblast transformation, as well as alveolar epithelial to mesenchymal transition and mouse lung fibrosis.8–10 Although there is a wide range of evidence indicating a correlation between KL-6 serum levels and the progression and severity of IPF, there is a lack of evidence regarding the biological activation and cell signalling of the MUC1 CT in IPF.
Following proteolysis of MUC1 at the SEA domain, MUC1-N is released and the C-terminal subunit (MUC1-C or β-chain) anchors the mucin to the surface of the cell.11 MUC1-C is short, consisting of a 58-amino acid extracellular domain, a 28-amino acid transmembrane domain and a 72-amino acid CT.12 The highly conserved CT in the C-terminal subunit of MUC1 modulates multiple intracellular signals13 implicated in different cellular processes, including adherens junction maintenance and morphogenesis.14 Several fibrotic and remodelling signals, such as β-catenin, platelet-derived growth factor receptor, fibroblast growth factor receptor (FGFR), epidermal growth factor receptor (EGFR) and mesenchymal epithelial transition (MET) tyrosine kinase receptor, can interact with MUC1-CT, thus exacerbating various cell transformation disorders.15
In this study, we analysed the previously unexplored role of MUC1-CT signal transduction in IPF, as well as the interaction between the transforming growth factor (TGF)-β1 signalling pathway and MUC1-CT, as promising drug targets in IPF. Using lung tissue, primary ATII cells, lung fibroblasts from patients with IPF and MUC1-knockout (MUC1-KO) mice, we report a a role for MUC1-CT in IPF, and provide mechanistic insight into the interactions between MUC1-CT, TGF-β1 and galectin-3.
Materials and methods
IPF lung tissue was obtained from patients undergoing surgery for organ transplantation (n=22). Lung explant control samples were obtained from the organ transplant programme of the University General Hospital of Valencia, Spain (n=21).
IPF was diagnosed according to the American Thoracic Society/European Respiratory Society consensus criteria.16 Informed written consent was obtained from all participants. Clinical data are provided in online supplementary table S1.
Supplemental material
Detailed descriptions of additional materials and methods are provided in the online data supplement.
Results
MUC1-CT is overexpressed and activated in the lungs of patients with IPF
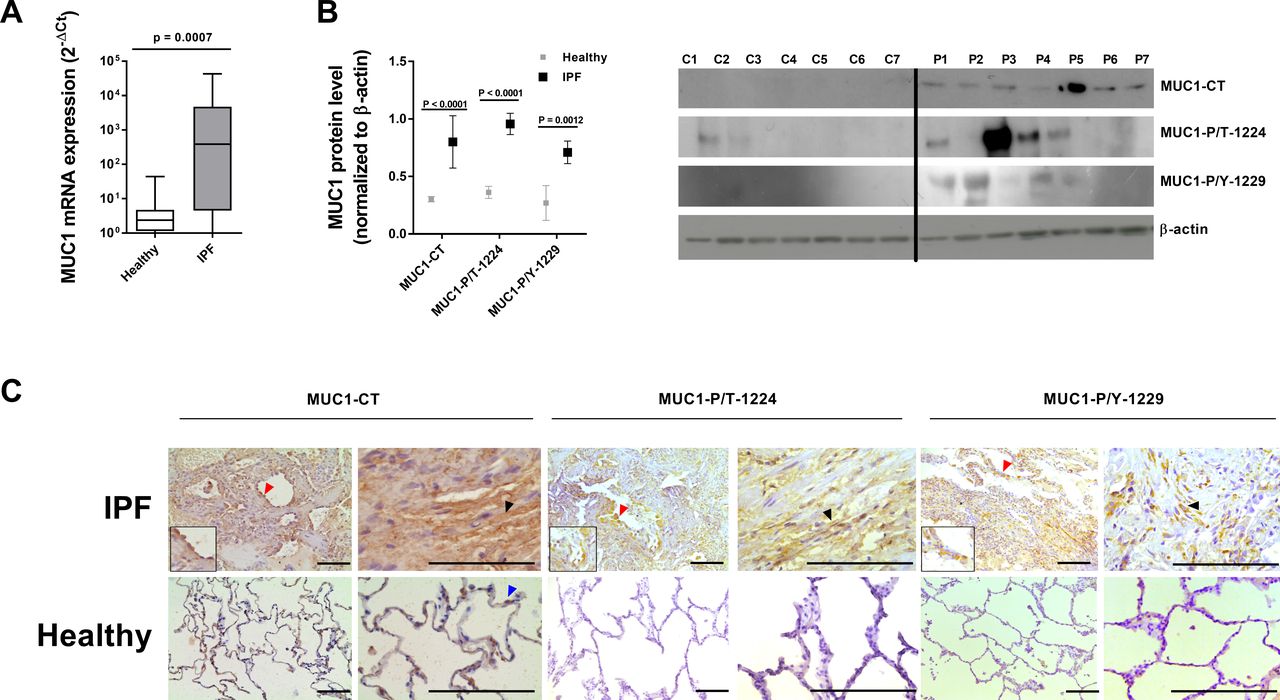
MUC1 transcript and MUC1-CT protein expression levels were elevated in lung tissue from patients with IPF compared with controls (figure 1A). In addition, whereas the active forms of MUC1-CT phosphorylated at Thr41 (CT aa position) (P/T 1224 aa position in full-length MUC1) and Tyr46 (CT aa position) (P/Y 1229 aa position in full-length MUC1) were upregulated in IPF lung tissue, the levels were low in control lung tissue (figure 1B). Immunohistochemistry of control lung sections showed weak MUC1-CT expression that was localised mainly in ATII cells (figure 1C). In contrast, in IPF lung tissue, MUC1-CT was elevated in hyperplastic ATII cells (figure 1C, red arrows) and fibroblasts (figure 1C, black arrows), and was mainly localised in the membrane/cytoplasm and nucleus. No phosphorylation of MUC1-CT at Thr41(1224) and Tyr46(1229) residues was detected in healthy lung tissue, but a similar distribution pattern to MUC1-CT was seen, including cytoplasmic and nuclear localisation in fibrotic areas of the lung. MUC1-CT overexpression and phosphorylation co-occurred in IPF lung tissue (online supplementary figure S1).

Mucin 1-cytoplasmic tail (MUC1-CT), and its phosphorylated forms MUC1-P/T-1224 and MUC1-P/Y-1229, are overexpressed in lung tissue from patients with idiopathic pulmonary fibrosis (IPF). Lung tissue was obtained from healthy controls (n=21) and patients with IPF (n=22). (A) MUC1 mRNA expression was analysed by real-time PCR. (B) MUC1-CT, MUC1-P/T-1224 and MUC1-P/Y-1229 protein expression levels were analysed by western blot analysis (n=14). (C) Immunohistochemistry of MUC1-CT, MUC1-P/T-1224 and MUC1-P/Y-1229. Scale bar: 100 µm. Red arrows indicate hyperplastic alveolar type II (ATII) cells. Black arrows indicate lung fibroblasts. Blue arrows indicate normal ATII cells. Data are shown as the ratio compared with β-actin for protein expression and 2−ΔCt for mRNA levels. Data are presented as box and whisker plots with median, IQR and minimum and maximum values. P values are based on the Mann–Whitney U test.
TGF-β1 collaborates with MUC1-CT to induce the ATII to mesenchymal and fibroblast to myofibroblast transitions
In primary ATII cells and the bronchoalveolar cell line A549, TGF-β1 significantly increased MUC1-CT Thr41(1224) and Tyr46(1229) phosphorylation after 40 min of stimulation (figure 2A) without an increase in MUC1-CT expression. TGF-β1 promoted the ATII to mesenchymal transition in A549 cells (figure 2B) and primary ATII cells (figure 2C), increasing the protein and gene expression of the mesenchymal markers alpha sooth muscle actin (α-SMA), collagen type I, snail family transcriptional repressor 2 (SLUG) and SNAIL, and decreasing the expression of the epithelial marker E-cadherin after 72 hours of stimulation (figure 2B–D). TGF-β1 increased the canonical Smad3 phosphorylation pathway, as well the non-canonical ERK1/2 phosphorylation and active-β-catenin pathways (figure 2B). In A549 cells transiently transfected with siRNA-MUC1, TGF-β1 did not induce the ATII to mesenchymal transition, nor activate β-catenin, but the Smad3 and ERK1/2 phosphorylation pathways were activated (figure 2B and D).
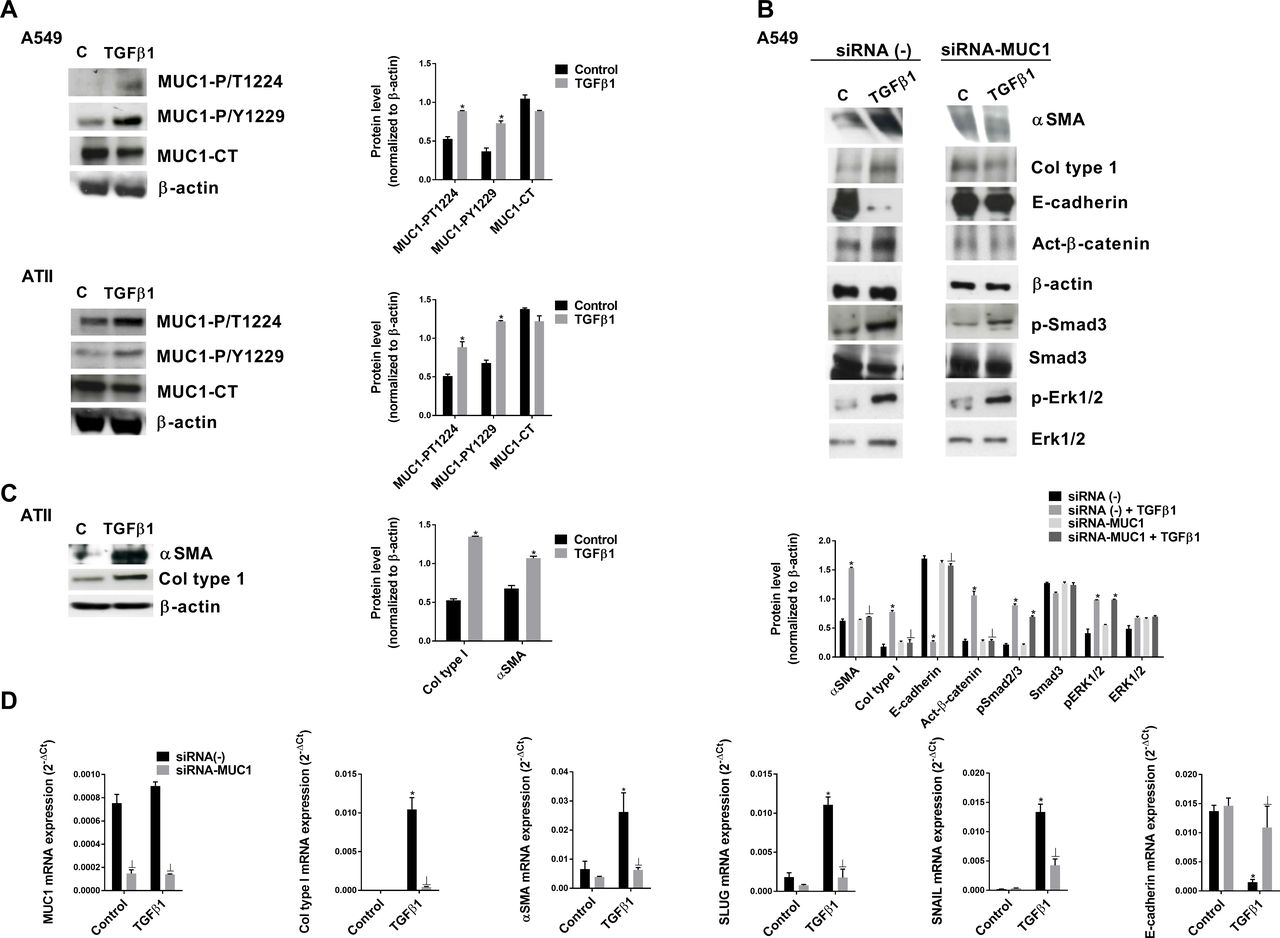
Transforming growth factor (TGF)-β1 and mucin 1-cytoplasmic tail (MUC1-CT) collaborate to induce the alveolar type II (ATII) to mesenchymal transition. Primary ATII cells were isolated from the lungs of control subjects. (A) A549 cells and ATII cells were stimulated for 40 min with 5 ng/mL TGF-β1, and MUC1-P/T-1224 and MUC1-P/Y-1229 expression were measured by western blot analysis. (B) The A549 cell line transfected with control siRNA(−) or siRNA-MUC1 was stimulated for 48 hours with 5 ng/mL TGF-β1 to measure α-SMA, collagen type I and E-cadherin, and for 40 min to measure active (act)-β-catenin, p-Smad3, Smad3, p-Erk1/2 and Erk1/2 by western blot analysis; quantification was performed by densitometry. (C) primary ATII cells stimulated for 72 hours with 5 ng/mL TGF-β1. (D) The A549 cell line transfected with control siRNA(−) or siRNA-MUC1 was stimulated for 48 hours with 5 ng/mL TGF-β1 to measure MUC1, collagen type I, α-SMA, SLUG, SNAIL and E-cadherin by quantitative PCR. Data are expressed relative to β-actin protein, and to 2−ΔCt for mRNA levels. The results are expressed as means±SE. One-way analysis of variance (ANOVA) (for A549 cells, three independent experiments performed in triplicate) or two-way ANOVA (for primary ATII cells, n=3 patients performed in triplicate) was followed by the post hoc Bonferroni test. *P<0.05 vs control; ⊥p<0.05 vs siRNA(−)+TGF-β1.
Similar results were observed in primary lung fibroblasts and the MRC5 lung fibroblast cell line (figure 3). TGF-β1 phosphorylated MUC1-CT at the Thr41(1224) and Tyr46(1229) residues (figure 3A), and induced the fibroblast to myofibroblast transition in MRC5 lung fibroblasts (Figure B) and primary fibroblasts (figure 3C) without an increase in MUC1-CT expression. In MRC5 cells transiently transfected with siRNA-MUC1, TGF-β1 did not induce the fibroblast to myofibroblast transition or activate β-catenin, but increased Smad3 expression and ERK1/2 phosphorylation (figure 3B and D).
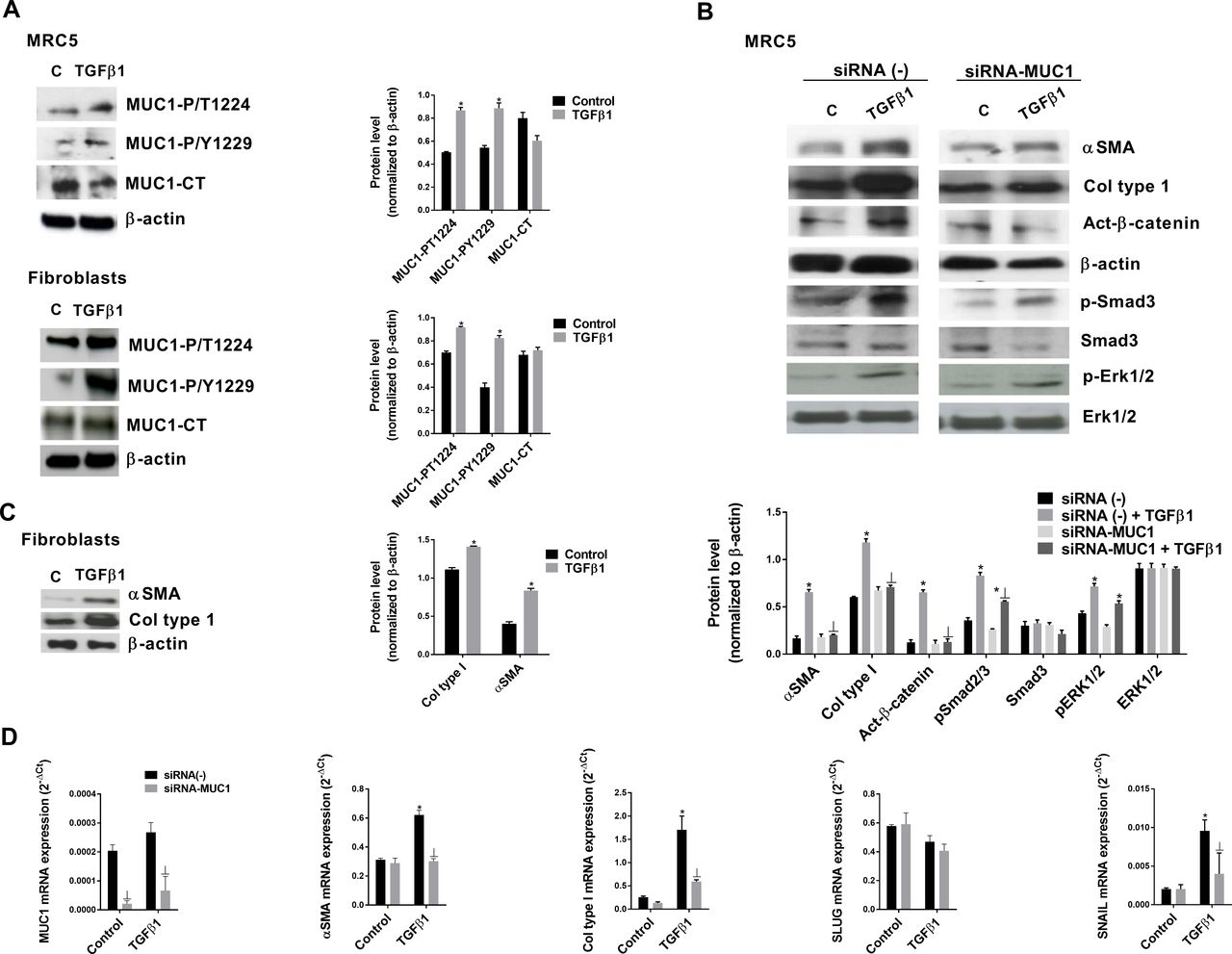
Transforming growth factor (TGF)-β1 and mucin 1-cytoplasmic tail (MUC1-CT) cooperate to induce the fibroblast to myofibroblast transition. Primary lung fibroblasts were isolated from the lungs of patients with idiopathic pulmonary fibrosis (IPF). (A) The MRC5 cell line and primary lung fibroblasts were stimulated for 40 min with 5 ng/mL TGF-β1, and MUC1-P/T-1224 and MUC1-P/Y-1229 expression levels were measured by western blot analysis. (B) The MRC5 cell line transfected with control siRNA(−) or siRNA-MUC1 was stimulated for 48 hours with 5 ng/mL TGF-β1 to measure α-SMA and collagen type I expression levels, and for 40 min to measure act-β-catenin, p-Smad3, Smad3, p-Erk1/2 and ERK1/2 expression levels by western blot analysis; quantification was performed by densitometry. (C) Primary lung fibroblast cells stimulated for 72 hours with 5 ng/mL TGF-β1. (D) The MRC5 cell line transfected with control siRNA(−) or siRNA-MUC1 was stimulated for 48 hours with 5 ng/mL TGF-β1 to measure MUC1, collagen type I, α-SMA, SLUG and SNAIL by quantitative PCR. Data are expressed relative to β-actin protein, and to 2−ΔCt for mRNA levels. The results are expressed as means±SE. One-way analysis of variance (ANOVA) (for MRC5 cells, three independent experiments performed in triplicate) or two-way ANOVA (primary fibroblasts, n=3 patients performed in triplicate) was followed by the post hoc Bonferroni test. *P<0.05 vs control; ⊥p<0.05 vs siRNA(−)+TGF-β1.
TGF-β1 activates Smad3 phosphorylation to promote phospho-Smad3/MUC1-CT and MUC1-CT/β-catenin nuclear protein complex and Smad-binding element activation
In ATII cells, TGF-β1 stimulus promoted the formation of a nuclear protein complex including phospho-Smad3/MUC1-CT and MUC1-CT/β-catenin, as demonstrated by co-localisation of phospho-Smad3/MUC1-CT and active-β-catenin/MUC1-CT, shown by immunoprecipitation (figure 4A) and confocal microscopy studies (figure 4B). TGF-β1 induced Smad-binding element (SBE) activation in A549 cells, which was attenuated in cells transiently transfected with siRNA-MUC1 (figure 4C). TGF-β1 stimulus also promoted nuclear protein complex formation of phospho-Smad3/MUC1-CT and MUC1-CT/β-catenin in primary lung fibroblasts from patients with IPF, as shown by immunoprecipitation and immunofluorescence confocal microscopy studies (figure 5A and B). MUC1-CT and β-catenin were localised on the cell membrane of ATII cells in lung sections from healthy patients, but phospho-Smad3 was absent, as indicated by fluorescence images (figure 5C). The expression levels of MUC1-CT and phospho-Smad3 were increased and co-localised in the cell cytoplasm and nuclei of lung fibrotic areas in patients with IPF. A similar distribution pattern was also observed with respect to MUC1-CT and β-catenin co-localisation in lung sections of patients with IPF (figure 5C).
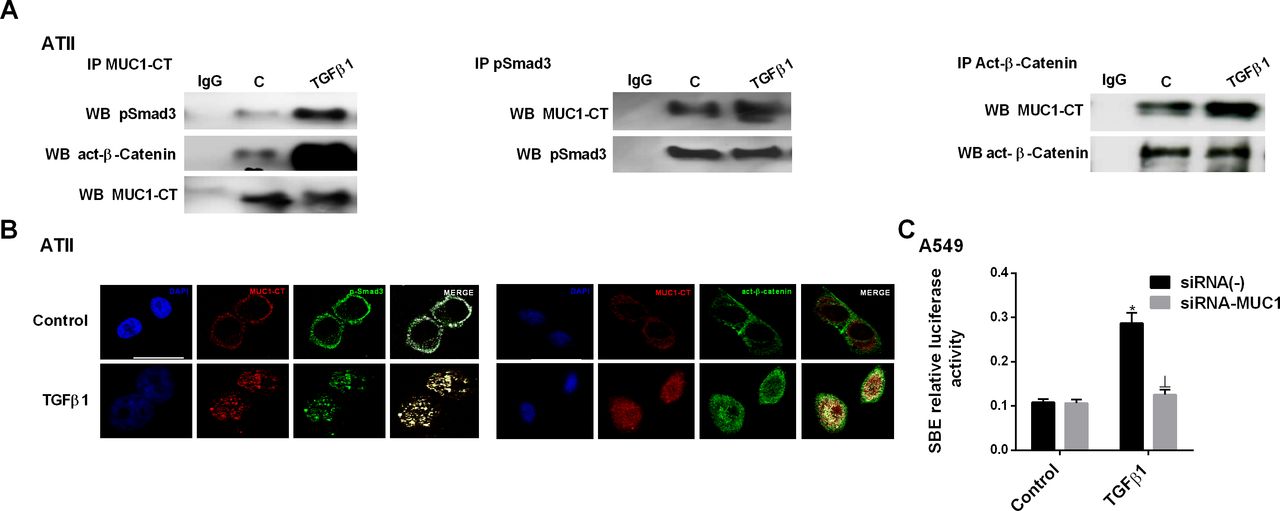
Mucin 1-cytoplasmic tail (MUC1-CT) co-localises with pSmad3 and active-β-catenin in the nuclei of alveolar type II (ATII) cells stimulated with transforming growth factor (TGF)-β1. ATII cells were stimulated with 5 ng/mL TGF-β1 for 1 hour. (A) Total protein was extracted and immunoprecipitated with MUC1-CT, phospho-Smad3 or act-β-catenin. Different western blot analyses were probed with antibodies to act-β-catenin, phospho-Smad3 and MUC1-CT (representative images are shown). Non-specific IgG was used as a negative isotype control for immunoprecipitation. (B) Co-localisation was analysed by confocal microscopy to generate a two-dimensional cytofluorogram that selected common localised points of both antibodies (white colour). Scale bars: 10 µm. Confocal immunofluorescence micrographs showed the nuclear translocation and co-localisation of MUC1-CT/act-β-catenin and MUC1-CT/phospho-Smad3. (C) In A549 cells, TGF-β1 induced Smad-binding element (SBE) activation, which was prevented in cells transiently transfected with siRNA-MUC1. The results are expressed as means±SE of three independent experiments performed in triplicate. One-way analysis of variance was followed by the post hoc Bonferroni test. *P<0.05 vs control; ⊥p<0.05 vs siRNA(−)+TGF-β1.

Mucin 1-cytoplasmic tail (MUC1-CT) co-localises with pSmad3 and act-β-catenin in the nuclei of lung fibroblasts stimulated with transforming growth factor (TGF)-β1, and in idiopathic pulmonary fibrosis (IPF) lung tissue. Primary lung fibroblasts from patients with IPF were stimulated with 5 ng/mL transforming growth factor (TGF)-β1 for 1 hour. (A) Total protein was extracted and immunoprecipitated with MUC1-CT, phospho-Smad3 or act-β-catenin. Different western blot analyses were probed with antibodies to active act-β-catenin, phospho-Smad3 and MUC1-CT (representative images are shown). Non-specific IgG was used as a negative isotype control for immunoprecipitation. (B) Co-localisation was analysed by confocal microscopy to generate a two-dimensional cytofluorogram that selected common localised points of both antibodies (white). Scale bars: 10 µm. Confocal immunofluorescence micrographs showed the nuclear translocation and co-localisation of MUC1-CT/act-β-catenin and MUC1-CT/phospho-Smad3. (C) Confocal immunofluorescence micrographs: co-localisation images of lung sections from healthy patients and patients with IPF probed with antibodies to MUC1-CT/act-β-catenin and MUC1-CT/phospho-Smad3.
The incubation of primary ATII cells and A549 cells, as well as primary lung and MRC5 fibroblasts, with the inhibitor of Smad3 phosphorylation, SIS3, significantly reduced TGF-β1-induced MUC1-CT phosphorylation at Thr41(1224) and Tyr46(1229) residues, suggesting a role of Smad3 activation in MUC1-CT phosphorylation (online supplementary figure S2). SIS3 also inhibited Smad3 phosphorylation and the increase in active β-catenin expression (online supplementary figure S2).
MUC1 mediates cell senescence and proliferation induced by TGF-β1 in ATII cells and lung fibroblasts
TGF-β1 has been shown to promote cell senescence in ATII cells and lung fibroblasts, as well as proliferation of lung fibroblasts, depending on the cell phenotype and cell culture conditions.17–19 In this study, we observed a critical threshold for IPF primary lung fibroblast phenotype changes after in vitro TGF-β1 stimulation. TGF-β1 at a dose of 10 ng/mL increased cell proliferation after 48 hours of stimulation (online supplementary figure S3A). At this point, lung fibroblasts showed increased β-galactosidase activity, thus indicating a change from active proliferative to senescent fibroblasts (online supplementary figure S3A–C). Proliferative and senescent fibroblast phenotypes were inhibited in siRNA-MUC1-transfected cells. In other experiments, we observed that TGF-β1 increased the β-galactosidase activity in A549 cells that was inhibited in siRNA-MUC1-transfected cells (online supplementary figure S3D).
Galectin-3 activates MUC1-CT independent of the TGF-β1 pathway and potentiates the effect of TGF-β1 on MUC1-CT phosphorylation
Galectin-3 is a recognised profibrotic factor in IPF20 and has been shown to activate MUC1 downstream signalling.21 Furthermore, galectin-3 prolongs TGF-β receptor signalling to Smads, activating β-catenin and thus resulting in concomitant nuclear accumulation of Smads and β-catenin, in turn leading to the transcription of mesenchymal genes in ATII cells.20 In the present study, the A549 cell line was transiently transfected to silence TβRI and TβRII using siRNA-TβRI/II. In control siRNA(−) cells, TGF-β1 increased Smad3 phosphorylation, and active-β-catenin and MUC1-CT phosphorylation at Thr41(1224) and Tyr46(1229) residues, effects that were significantly potentiated by the addition of galectin-3 (online supplementary figure S4). Galectin-3 stimulus in the absence of TGF-β1 also increased phospho-Smad3, MUC1-CT Thr41(1224) and Tyr46(1229) phosphorylation, and active-β-catenin, similar to TGF-β1 stimulus. The effects of TGF-β1 were suppressed in cells transiently transfected with siRNA-TβRI/II. Galectin-3, or a combined TGF-β1/galectin-3 stimulus, did not modify Smad3 phosphorylation in siRNA-TβRI/II cells, but increased the expression of active-β-catenin and MUC1-CT Thr41(1224) and Tyr46(1229) phosphorylation (online supplementary figure S4), suggesting that galectin-3 can stimulate MUC1-CT in a manner both dependent and independent of the activation of TGF-β receptors.
MUC1-CT CQC motif inhibition reduces TGF-β1-induced MUC1-CT nuclear translocation, the ATII to mesenchymal transition, fibroblast to myofibroblast transition and bleomycin-induced mouse lung fibrosis
GO-201, an inhibitor of the CQC motif of MUC1-CT, inhibited the effect of TGF-β1 on the increase in mRNA expression of the mesenchymal markers, α-SMA and vimentin, as well as the decrease in E-cadherin protein expression (figure 6A–C). The gene expression levels of the cell senescence markers, p21 and p16, were elevated after TGF-β1 stimulation and inhibited by GO-201 in ATII cells (figure 6D and E). In lung fibroblasts from patients with IPF, TGF-β1 increased gene expression of the myofibroblast marker collagen type I, as well as the senescence markers, p21 and p16, which were inhibited by GO-201 (figure 6F–6H). GO-201 inhibited the fibroblast proliferation induced by TGF-β1 after 48 hours of stimulation (figure 6I) as well as the activation of SBE in A549 cells stimulated with TGF-β1 (figure 6J). In ATII cells, TGF-β1 promoted the nuclear localisation of MUC1-CT after 1 hour of stimulation, which was effectively blocked by GO-201, as shown by immunofluorescence analysis (figure 6K).

Inhibition of mucin 1 (MUC1) nuclear translocation suppressed the alveolar type II (ATII) to mesenchymal and fibroblast to myofibroblast transitions. Human ATII primary cells were isolated from the lungs of control subjects, and primary human lung fibroblasts were isolated from the lungs of patients with idiopathic pulmonary fibrosis (IPF). Human ATII primary cells (A–E) and primary human lung fibroblasts (F–H) were stimulated for 48 hours with 5 ng/mL transforming growth factor (TGF)-β1 in the presence or absence of 5 µM GO-201. Total protein and RNA from cell lysates were analysed by quantitative PCR (qPCR) and western blot analysis. (A) Mesenchymal α-SMA, (B) vimentin and (F) collagen type I were measured by qPCR and (C) epithelial E-cadherin was measured by western blot analysis. (D, G) Expression levels of the senescence markers p21 and (E, H) p16 were measured by qPCR. (I) MRC5 proliferation for 48 hours was evaluated by BrdU assay. GO-201 was added at 5 µM 30 min before 10% fetal bovine serum and 5 ng/mL TGF-β1, and remained for 48 hours. (J) In A549 cells, TGF-β1 induced Smad-binding element (SBE) activation, which was prevented in cells treated with GO-201 5 µM. (K) ATII cells were fixed, permeabilised and used for MUC1-CT (red) immunostaining after stimulation for 1 hour with 5 ng/mL TGF-β1 in the presence or absence of 5 µM GO-201. Nuclei were stained blue with DAPI. Scale bars: 10 µm. Results are expressed as means±SE of three patients or three independent experiments performed in triplicate. Two-way analysis of variance was followed by the post hoc Bonferroni test. *P<0.05 vs control; #p<0.05 vs TGF-β1.
In other experiments, GO-201 was administered at a dose of 15 mg/kg/daily, as part of a therapeutic protocol from day 7 to day 14 after a single dose of bleomycin. Lung collagen deposition was reduced in mice treated with GO-201, as indicated by the reduction in Ashcroft score and hydroxyproline level (online supplementary figure S5A,B). The administration of GO-201 also decreased the mRNA expression of profibrotic markers, such as collagen type I, TGF-β1, connecting tissue growth factor (CTGF) and interleukin (IL)-6 (online supplementary figure S5C). The activation of downstream signals of TGF-β1, such as Smad3 and Erk1/2 phosphorylation, as well as the activation of β-catenin, was decreased after GO-201 administration (online supplementary figure S5D), as observed for the senescence marker, p21 (online supplementary figure S5D). In lung tissue, bleomycin administration promoted the MUC1-CT nuclear localisation that was effectively blocked by GO-201 (online supplementary figure S5E).
Lung fibrosis induced by bleomycin is reduced in MUC1-KO mice
Intratracheal (IT) bleomycin administration induced robust lung fibrosis that increased the mortality of mice by ~60% after 14 days (figure 7A). Mortality was improved in MUC1-KO mice to ~30% (figure 7A), while wild-type (WT) control and MUC1-KO control mice showed 100% survival. Bleomycin-treated WT animals showed increased Penh values; these were significantly lower in the MUC1-KO bleomycin group (figure 7B), suggesting improvement of lung function. However, Penh value is not always correlated with pulmonary resistance, so the results should be interpreted with caution.22 Bleomycin-treated WT mice showed higher Ashcroft scores (collagen deposition) and reduced air spaces (higher micro-CT Hounsfield units) than MUC1-KO mice, reflecting a reduction in the lung fibrotic extension.
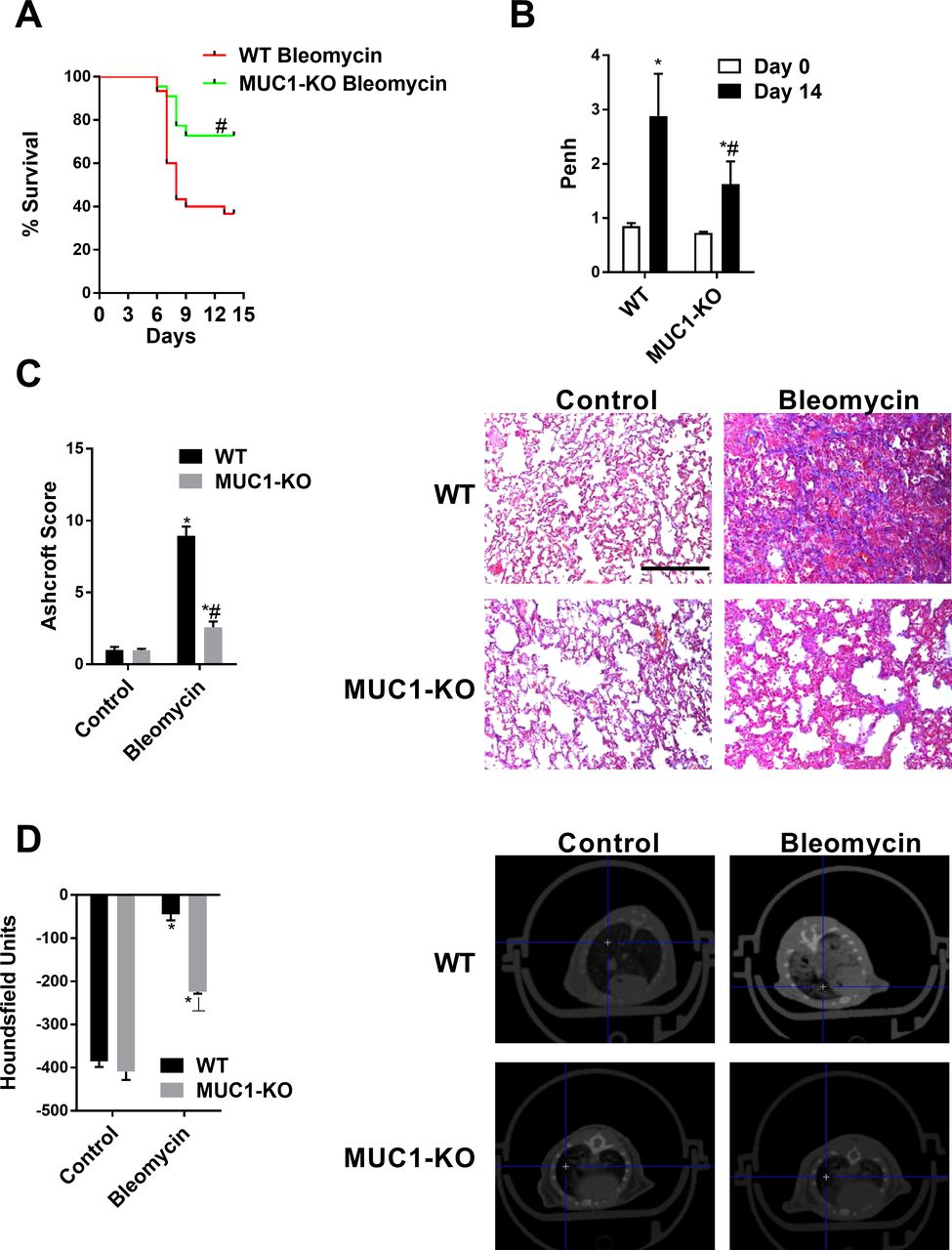
Mucin 1 (MUC1)-knockout (KO) improves survival and inhibits bleomycin-induced lung fibrosis. Wild-type (WT) C57BL/6 mice and MUC1-KO C57BL/6 mice received a single intratracheal dose of bleomycin (1.5 U/kg) on day 1 (n=10). (A) Kaplan-Meier survival analysis of mice treated with bleomycin for 14 days. (B) Pulmonary functions were evaluated as enhanced respiratory pause (Penh) on day 14 after bleomycin administration. Whole-body plethysmography was performed and Penh was used as a non-invasive index of airway dysfunction. (C) Masson’s trichrome staining (right panels, scale bars: 100 µm) of WT control, WT bleomycin, MUC1-KO control and MUC1-KO bleomycin tissue. Ashcroft fibrosis scores were calculated as described in the ‘Materials and methods’ section. One-way analysis of variance (ANOVA) was followed by the post hoc Bonferroni test. *P<0.05 vs day 0 or control; #p<0.05 vs WT. (D) Micro-CT images were acquired on day 14 and quantified as Hounsfield units. The results are expressed as means±SE (10 mice per group). One-way ANOVA was followed by the post hoc Bonferroni test. *P<0.05 vs control; #p<0.05 vs WT.
IT bleomycin increased right ventricle hypertrophy (online supplementary figure S6A) and impaired the pulmonary circulation, as indexed by the decreased micro-SPECT-albumin macroaggregate-Tc99m radiation signal (online supplementary figure S6B and D). These values were lower in bleomycin MUC1-KO mice (online supplementary figure S6B and D). Lung metabolic activity was also increased in bleomycin-treated WT mice with higher lung standardised uptake values of micro-18F fluorodeoxyglucose-positron emission tomography compared with bleomycin MUC1-KO mice (online supplementary figure S6C and E).
The IT bleomycin administration in WT mice produced greater numbers of inflammatory cells in BALF than seen in MUC1-KO mice (online supplementary figure S7A,B). The levels of TGF-β1 and IL-6 in the BALF, as well as fibrotic marker gene mRNA expression in inflammatory cells in BALF, were higher in bleomycin-treated WT mice than in MUC1-KO mice (online supplementary figure S7C–F). WT and MUC1-KO control mice showed similar levels of BALF inflammatory cells and TGF-β1/IL-6.
The mRNA gene and protein expression of recognised fibrotic markers and mediators, such as collagen type I, TGF-β1, CTGF, IL-6 and IL-13 were induced by bleomycin in lung tissue from WT mice, while MUC1-KO animals showed minimal elevation (online supplementary figure S8A, B). The expression of MUC1-CT and its phosphorylated forms at Thr41(1224) and Tyr46(1229) were increased in lung tissue from WT mice after bleomycin administration. Furthermore, the activation of downstream signals of TGF-β1, such as Smad3 and Erk1/2 phosphorylation, as well as the activation of β-catenin, were increased in bleomycin-treated WT mice, while MUC1-KO mice showed levels similar to those in untreated mice (online supplementary figure S8B). The expression level of the senescence marker, p21, was also increased in bleomycin-treated WT mice, but not in MUC1-KO mice.
Immunohistochemical analysis showed elevated expression of TGF-β1 and NOX4 in WT mice exposed to bleomycin, mainly in pleural epithelial cells, ATII cells and fibroblasts, but not in MUC1-KO mice. Furthermore, MUC5B mucin expression, which is altered in IPF,23 was increased in bleomycin-treated WT mice and distributed in altered honeycomb lesions in the lung, as observed for MUC1-CT overexpression (online supplementary figure S9). Immunofluorescence confocal micrographs of mouse lung sections showed co-localisation of β-catenin/MUC1-CT in the plasma membrane of ATII cells from WT control mice, and cytoplasmic and nuclear distribution of β-catenin/MUC1-CT in bleomycin-treated WT mice (online supplementary figure S10). This distribution pattern was not observed in MUC1-KO mice. Similar distribution patterns were observed for MUC1-CT and phosphorylated Smad3, which were co-localised in the cytoplasm and nuclei of fibrotic areas in bleomycin-treated WT mice (online supplementary figure S10).
Discussion
The present study provided novel evidence of intracellular bioactivation of MUC1-CT in IPF, which may be of value to understand the pathogenesis of IPF, as well as to identify future drug targets.
MUC1 mRNA expression and KL6/MUC1 protein expression were shown previously to be elevated in human lung tissue from patients with various interstitial lung diseases compared with healthy controls.7 24 The distribution of KL6/MUC1 was found to be restricted to the ATII cell surface in healthy patients and patients with IPF.7 Under conditions of lung fibrosis, elevated levels of metalloproteases mediate proteolytic cleavage close to the plasma membrane, thus releasing the extracellular KL6/MUC1 domain that can be detected in many biological fluids. Soluble KL6/MUC1 has activity in lung fibroblasts, where it promotes cell proliferation, migration and myofibroblast transition, activating unknown targets8 9; antibodies against KL6 were shown to attenuate bleomycin-induced lung fibrosis.10 However, some contradictory findings have also been reported in the literature, as silica-induced lung fibrosis in MUC1-KO mice was shown to increase, rather than decrease, lung fibrosis.25
Currently, there are no data regarding the expression, location and function of intracellular MUC1-CT in IPF. In this study, MUC1-CT was increased in lung tissue from patients with IPF, but not in the lungs of healthy subjects, and was located mostly in hyperplastic ATII cells and fibroblasts in fibrotic areas, mainly distributed in the cytoplasm and nucleus. MUC1-CT phosphorylated at Thr41(1224) and Tyr46(1229) was detected only in IPF lung tissue, with a distribution pattern similar to non-phosphorylated MUC1-CT; this suggests a different role to that of the previously studied KL6/MUC1. Although MUC1-CT expression was increased overall in patients with IPF, there was variability among individuals, and between phosphorylation states within individuals, as detected by western blot analysis; this may reflect the heterogeneity of IPF disease.
MUC1-CT consists of 72 amino acids, with 13 documented tyrosine and serine/threonine phosphorylation sites, which contribute to interactions with various effectors linked to cell transformation.11 Thr41(1224) phosphorylation at the TDR amino acid site, and Tyr46(1229) phosphorylation at the YEKV amino acid site, induce binding of the MUC1-CT SAGNGGSSLS sequence to β-catenin, thereby promoting the activation of Wnt target genes26 and CTGF,27 both of which are associated with IPF.5 28 Therefore, loss of MUC1-CT expression decreases the levels of total and active β-catenin. Furthermore, Thr41(1224) and Tyr46(1229) phosphorylation promote nuclear translocation of MUC1-CT/β-catenin complex to activate dependent gene expression.13 In addition, the activation of several growth factor receptors, including EGFR, FGFR3, platelet-derived growth factor B and MET, results in phosphorylation of MUC1-CT, which in turn modulates their signal transduction.11 Most of these growth factors participate in lung fibrosis,5 suggesting a possible role of MUC1-CT signalling in IPF.
In this study, we analysed the possible connection of TGF-β1 (the main profibrotic factor) with MUC1-CT. TGF-β1 phosphorylated Smad3, which promoted Thr41(1224) and Tyr46(1229) MUC1-CT phosphorylation in ATII cells and lung fibroblasts. The inhibition of phospho-Smad3 by SIS3 blocked TGF-β1-induced Thr41(1224) and Tyr46(1229) phosphorylation, indicating that phospho-Smad3 is required for phosphorylation of MUC1-CT. The bioactivation of MUC1-CT by phospho-Smad3 increased the active form of β-catenin, which was inhibited in siRNA-MUC1 cells, thus suggesting that Thr41(1224) and Tyr46(1229) MUC1-CT phosphorylation are needed to increase active-β-catenin in ATII cells and lung fibroblasts from patients with IPF, as demonstrated previously in other systems.13 Immunoprecipitation analysis revealed that MUC1-CT forms protein complexes with phospho-Smad3 and active-β-catenin in response to TGF-β1; this stimulus promoted the nuclear translocation of phospho-Smad3/MUC1-CT/β-catenin complex, which is required to activate the SBE DNA sequence to promote profibrotic gene expression. Nuclear co-localisation of the phospho-Smad3/MUC1-CT/β-catenin complex was also observed in lung tissue sections of patients with IPF, and in an animal model of bleomycin-induced lung fibrosis, thus reproducing the in vitro observations. Although we have not explored the interaction between phospho-Smad3 and β-catenin, previous studies have shown that TGF-β1 promotes the interaction of pSmad3 with active β-catenin, thus activating the SBE transcription element to induce the ATII to mesenchymal transition.29 The Thr41(1224) and Tyr46(1229) MUC1-CT phosphorylation, phospho-Smad3 and active-β-catenin were also increased in the lung tissue of the bleomycin-treated WT animal model, as were the levels of fibrotic mediators, such as TGF-β1, CTGF, collagen type I, IL-13 and IL-6, which were not increased in MUC1-KO mice. These results suggest a role of MUC1-CT in controlling fibrogenesis.
Lung cells in IPF undergo different types of transformation.3 The ATII to mesenchymal transition is observed in experimental models, and there is some support for a similar process in disease states in vivo, potentially contributing to the local myofibroblast population.3 The fibroblast to myofibroblast transformation has been also observed in IPF as a source of lung myofibroblasts, a cell phenotype with characteristics shared by both smooth muscle cells and fibroblasts that allows contraction and migration, as well as the release of extracellular matrix to invade lung tissue. TGF-β1 is the best characterised and most potent cellular transformer of fibroblasts and ATII cells, and multiple pathways can modulate its function.30 This study provided novel evidence of the interaction between the TGF-β1 canonical pathway and MUC1-CT bioactivation, which induces the ATII to mesenchymal and fibroblast to myofibroblast transitions. Neither A549 cells nor MRC5 fibroblasts transiently transfected with siRNA-MUC1 were transformed to myofibroblasts after TGF-β1 stimulation. Previous studies of cancer cells showed that MUC1-CT can induce the epithelial to mesenchymal transition and cellular invasion by a ZEB1-mediated mechanism, as well as mediating Wnt/β-catenin activation and Snail expression.31 32 In this study, MUC1-CT mediated the activation of β-catenin, as well as the expression of Snail in ATII cells and lung fibroblasts. Furthermore, the effects of MUC1-CT on the ATII to mesenchymal and fibroblast to myofibroblast transitions were dependent on MUC1-CT nuclear translocation. The MUC1-CT nuclear translocation is also mediated by a mechanism that is dependent on its oligomerisation, and thus involves interaction with importin β.11 Mucin dimerisation through the CQC motif (the first three aa of the MUC1-CT 72 aa region) yields a nuclear targeting signal based on the adjacent RRK motif, thus promoting MUC1-CT nuclear internalisation.33 In this regard, blocking the MUC1-CT CQC motif abrogates oligomerisation, and is therefore a target for pharmacological treatments to inhibit MUC1-CT nuclear localisation and transcriptional function. In this study, the MUC1-CT CQC motif inhibitor, GO-201, inhibited TGF-β1-induced MUC1-CT nuclear translocation, as well as the ATII to mesenchymal and fibroblast to myofibroblast transitions, thus demonstrating the modulatory role of MUC1-CT with respect to the effects of TGF-β1 on fibrotic cell transformation. This study did not provide direct evidence for TGF-β1-induced MUC1-CT oligomerisation. However, the TGF-β1-induced MUC1-CT nuclear localisation, and inhibition thereof by GO-201, indirectly confirmed this process. Similar findings were observed in the bleomycin-induced mouse lung fibrosis model, where GO-201 inhibited the deposition of collagen and fibrosis in the lungs.
In addition to cell transformation, senescence of ATII cells and fibroblasts are key pathological processes in IPF. The metabolically active, hypersecretory and apoptosis-resistant senescence phenotype of ATII cells and lung fibroblasts is abundant in the lungs of patients with IPF and contributes to the release of a number of fibrotic growth factors, such as TGF-β1, CTGF and IL-13.34 In addition, the proliferative fibroblast phenotype co-exists with the senescent phenotype, indicating complex interrelations of lung cells in IPF. In this study, TGF-β1 increased fibroblast proliferation; this was followed by cell senescence, as outlined previously.17 19 The inhibition of MUC1-CT expression via si-RNA-MUC1, or of MUC1-CT nuclear translocation using GO-201, blocked the ATII cell senescence, fibroblast senescence and lung fibroblast proliferation induced by TGF-β1. Similar results were observed in the MUC1-KO model, and in animals treated with GO-201, wherein p21 senescence marker levels were decreased. Although β-galactosidase staining has been used as a marker of cell senescence, this is a preliminary screening tool that does not provide confirmation of senescence. Alternative markers of senescence, such as p21, could provide additional information.
The inhibition of senescence could be explained by the interaction with, and activation of, β-catenin by MUC1-CT, as previous studies have suggested that Wnt/β-catenin acts as a driver of cell senescence in IPF.35 However, SBE activation by the phospho-Smad3 pathway also increases the expression of the senescence markers, p21 and p16,36 which link MUC1-CT with TGF-β1 canonical (Smad3) and non-canonical (β-catenin) pathways to mediate cell senescence. MUC1-CT also mediated the lung fibroblast proliferation induced by TGF-β1, but not via the ERK1/2 pathway as seen in cancer cells21; instead, it may have enhanced the SBE transcription expression of cyclin kinases.37
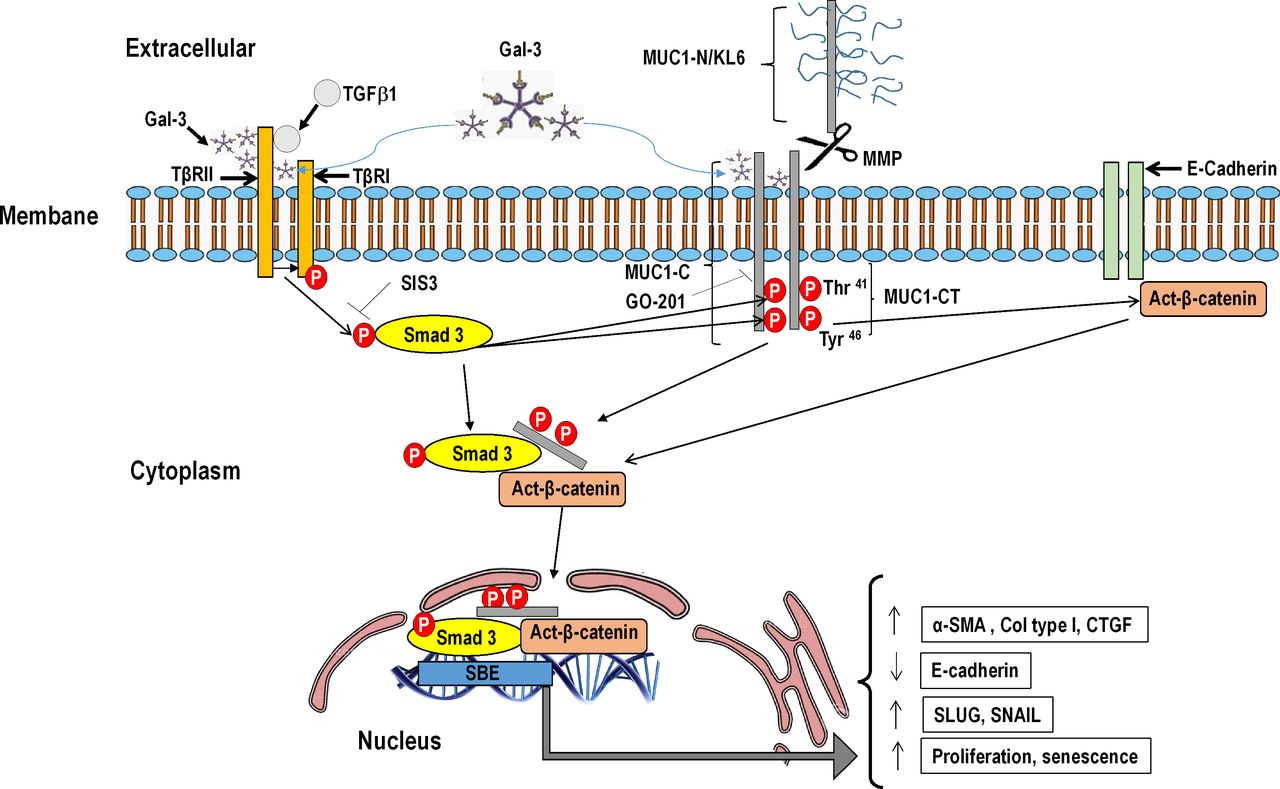
Galectin-3 is a promising target for treatment of fibrotic diseases, including IPF.1 Previous studies have demonstrated that galectin-3 prolongs TβR signalling, resulting in cell-surface retention and promoting Smad and AKT/β-catenin signalling in ATII cells, thus leading to cell transformation and lung fibrosis.20 In this study, we extended previous observations and demonstrated that galectin-3 reinforces TGF-β1 activation of Smad3, and MUC1-CT Thr41(1224) and Tyr46(1229) phosphorylation, thus increasing the expression of active-β-catenin. Furthermore, galectin-3 also has TβR-independent effects, directly activating MUC1-CT Thr41(1224) and Tyr46(1229) phosphorylation and active-β-catenin. These results indicated that MUC1-CT can be activated indirectly via phospho-Smad3, and directly via galectin 3, as outlined in another recent study.21 In IPF, increased metalloprotease levels mediate proteolytic cleavage close to the plasma membrane, thus releasing MUC1-N. The remaining MUC1-C includes a 58-amino acid extracellular domain that is glycosylated on Asn,36 and then serves as a binding site for the galectin-3 ligand that acts as a bridge between MUC1 and different growth factor receptors.38 This may be represented by TβR signalling, as shown in the present study (figure 8). However, these conclusions have been taken using indirect approaches and there is no evidence of the direct protein/protein interaction or complex formation between galectin-3, MUC1-CT and TβR, which guarantees future research.
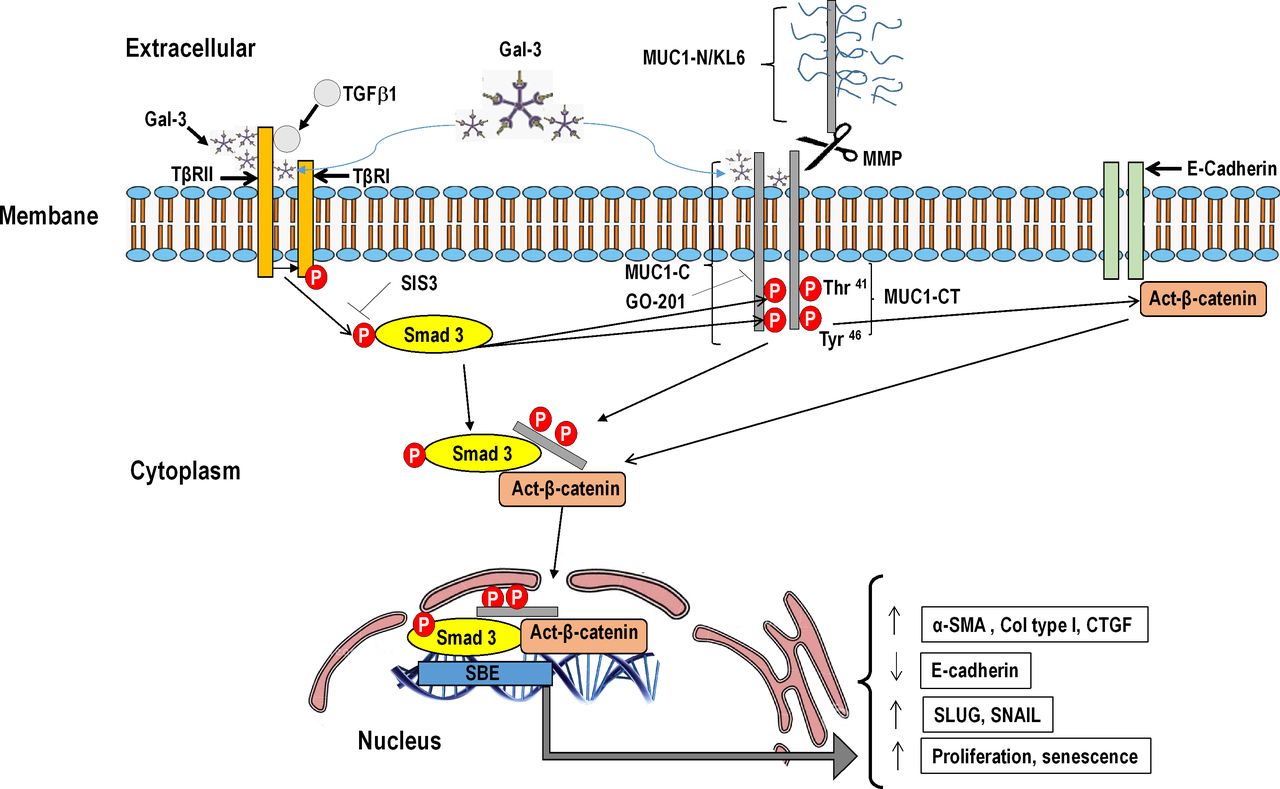
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic illustration showing novel evidence for bioactivation of mucin 1-cytoplasmic tail (MUC1-CT) in idiopathic pulmonary fibrosis (IPF). Under conditions of lung fibrosis, elevated levels of metalloproteases mediate proteolytic cleavage of MUC1 close to the plasma membrane, thus releasing extracellular KL6/MUC1 domain. MUC1-CT and its phosphorylated forms at Thr41(1224) and Tyr46(1229) are overexpressed in IPF lung tissue. Thr41(1224) and Tyr46(1229) MUC1-CT phosphorylation are induced by transforming growth factor (TGF)-β1. TGF-β1 binds to TβRI/II, leading to the recruitment and phosphorylation of Smad3. Phosphorylated Smad3 promotes the phosphorylation of MUC1-CT at Thr41 and Tyr46, thus increasing the active form of β-catenin. MUC1-CT forms protein complexes with phospho-Smad3 and act-β-catenin in response to TGF-β1; this stimulus promotes nuclear translocation of the phospho-Smad3/MUC1-CT/β-catenin complex, which is required to activate the Smad-binding element (SBE) DNA sequence and in turn promote profibrotic gene expression, proliferation or cell senescence. Galectin-3 (Gal-3) binds to TβRI/II and reinforces TGF-β1 activation of Smad3, and MUC1-CT Thr41(1224) and Tyr46(1229) phosphorylation, thus increasing the expression of act-β-catenin. Furthermore, Gal-3 binds to MUC1-CT, directly activating MUC1-CT Thr41(1224) and Tyr46(1229) phosphorylation, and act-β-catenin. Inhibition of phospho-Smad3 using SIS3 blocks TGF-β1-induced Thr41(1224) and Tyr46(1229) phosphorylation, thus reducing the amount of act-β-catenin and the nuclear translocation of the phospho-Smad3/MUC1-CT/β-catenin complex. The MUC1-CT nuclear translocation is also mediated by a mechanism that is dependent on its oligomerisation. In this regard, GO-201 blocks the MUC1-C CQC motif and abrogates oligomerisation, TGF-β1-induced MUC1-CT nuclear translocation and MUC1-CT transcriptional function.
In summary, the present study provided novel evidence for bioactivation of MUC1-CT in lung tissue and cells from patients with IPF, as well as in an animal model of lung fibrosis, which facilitate identification of new targets to treat this devastating disease.
Acknowledgments
The authors would like to thank Sonia Priego for assistance in analysing the confocal microscopy images at the Microscopy Service of the UCIM Faculty of Medicine, Valencia, Spain.
References
Footnotes
JM and BB contributed equally.
Contributors JM, BB, PM, AP-C, EM and JC designed and performed experiments and data analysis. JM, BB, JE, MA, EA and JC designed experiments and oversaw all data analysis. JM, BB, EM, JE, MA, EA and JC drafted the manuscript. All authors have critically revised the manuscript. All authors have read, reviewed and approved the final manuscript as submitted to take public responsibility for it.
Funding This work was supported by the grants SAF2017-82913-R (JC), FIS PI17/02158 (JM), JR18/00050 (JM), Fondo Europeo de desarrollo Regional (FEDER), RTI2018-096827-B-I00 (EM), CIBERES (CB06/06/0027), TRACE (TRA2009-0311), CM16/0022 (AP-C) of the Spanish Government and by research grants from the Regional Government Prometeo 2017/023/UV (JC, EM), ACIF/2016/341 (BB) from 'Generalitat Valenciana'.
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval The protocol was approved by the local research and independent ethics committee of the University General Consortium Hospital of Valencia, Spain (CEI CHGUV/052016).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.
Linked Articles
- Airwaves