Article Text

Abstract
Neutrophils represent a prominent source of pathology in an array of persistent pulmonary diseases. A recent article published in Science describes a novel anti-inflammatory pathway that degrades the neutrophil chemoattractant Pro-Gly-Pro (PGP) to limit neutrophilic inflammation of the lung. Degradation of PGP was mediated through the action of leukotriene A4 hydrolase (LTA4H), an enzyme classically recognised for its capacity to generate another neutrophil chemoattractant, leukotriene B4 (LTB4). The same enzyme therefore has opposing proinflammatory (LTB4 generation) and anti-inflammatory (PGP degradation) activities that govern neutrophilic inflammation. Intriguingly, cigarette smoke, a key risk factor for the development of chronic obstructive pulmonary disease, impedes PGP degradation but not LTB4 generation by LTA4H. Cigarette smoke therefore essentially converts LTA4H into an exclusively proinflammatory enzyme, whereby both PGP and LTB4 can drive persistent neutrophila observed in chronic obstructive pulmonary disease. In recent years there has been significant pharmaceutical interest in the development of LTA4H inhibitors to alleviate LTB4-mediated pathologies. In light of these new findings, such strategies should be viewed with caution since they may inadvertently prevent PGP degradation and promote chronic neutrophilic inflammation.
Statistics from Altmetric.com
Neutrophils are critical components of the body's immune response to infection, being readily mobilised to the site of infection and disposing of the invading pathogen with a potent arsenal of antimicrobial products. However, these same products are indiscriminate in toxicity and can cause significant bystander or ‘collateral’ damage to surrounding host tissue. Accordingly, neutrophils must be readily cleared from a site of infection, with persistent neutrophilia implicated in the pathology of chronic lung diseases such as chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF) and severe asthma. Anti-inflammatory steroids exhibit limited benefits in these diseases and have even been shown to promote neutrophil survival. There is therefore an urgent need to develop novel therapeutic strategies to alleviate neutrophil-mediated pathologies.
Signals that drive neutrophil recruitment and maintenance offer plausible therapeutic targets. Neutrophils are mobilised from the vasculature and into the lung in response to a broad array of chemoattractant signals. For a long time it has been known that fragments of structural proteins such as collagen and elastin that constitute the lung architecture can cause the recruitment of inflammatory cells. Proline-Glycine-Proline (PGP) is a peptide of just three amino acids, generated from collagen, that can recruit neutrophils by mimicking key sequences found in certain other neutrophil chemoattractants such as interleukin 8 (IL-8). PGP is generated from collagen by the sequential action of enzymes called matrix metalloproteinases followed by a secondary enzyme, prolyl endopeptidase.1 Neutrophils contain the full enzymatic repertoire required to generate PGP from collagen and are therefore capable of driving a self-sustained vicious circle of inflammation. Significant concentrations of PGP have been detected in chronic lung diseases such as COPD, CF and bronchiolitis obliterans syndrome, where they maintain neutrophilic inflammation at a time when other chemoattractant levels have subsided.1–3
A recent paper published in Science describes a novel anti-inflammatory pathway whereby PGP is degraded to switch off neutrophilic inflammation.4 Influenza infection of mice elicits acute pulmonary neutrophilic inflammation with concomitant release of PGP-generating enzymes but no PGP. Failure to detect PGP was found to be due to the activity of an enzyme being released by cells into the extracellular environment that could degrade this peptide. This enzyme was found to be leukotriene A4 hydrolase (LTA4H). LTA4H is classically recognised for a secondary activity that resides inside cells, where it converts leukotriene A4 (LTA4) into leukotriene B4 (LTB4). LTB4 is an extremely proinflammatory mediator, capable of recruiting and activating an array of immune cells including neutrophils and implicated in the pathologies of acute and chronic diseases. Thus, LTA4H exhibits opposing proinflammatory (LTB4 generation) and anti-inflammatory (PGP degradation) roles that govern neutrophil recruitment. Lung epithelial cells and neutrophils were shown to be capable of releasing extracellular LTA4H. The release by neutrophils suggests that the same cells normally coordinate the release of PGP-generating and PGP-degrading enzymes in order to resolve neutrophilic inflammation and limit tissue damage (figure 1A).
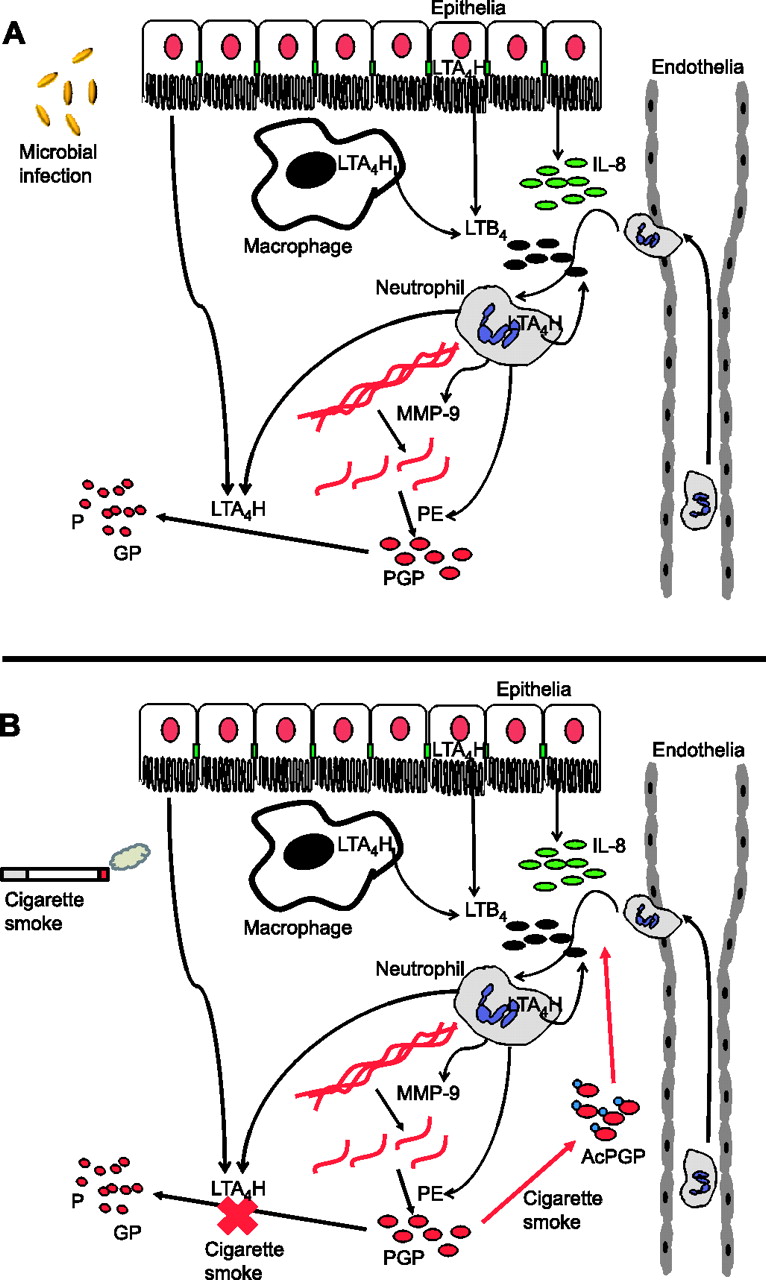
{kind=link}
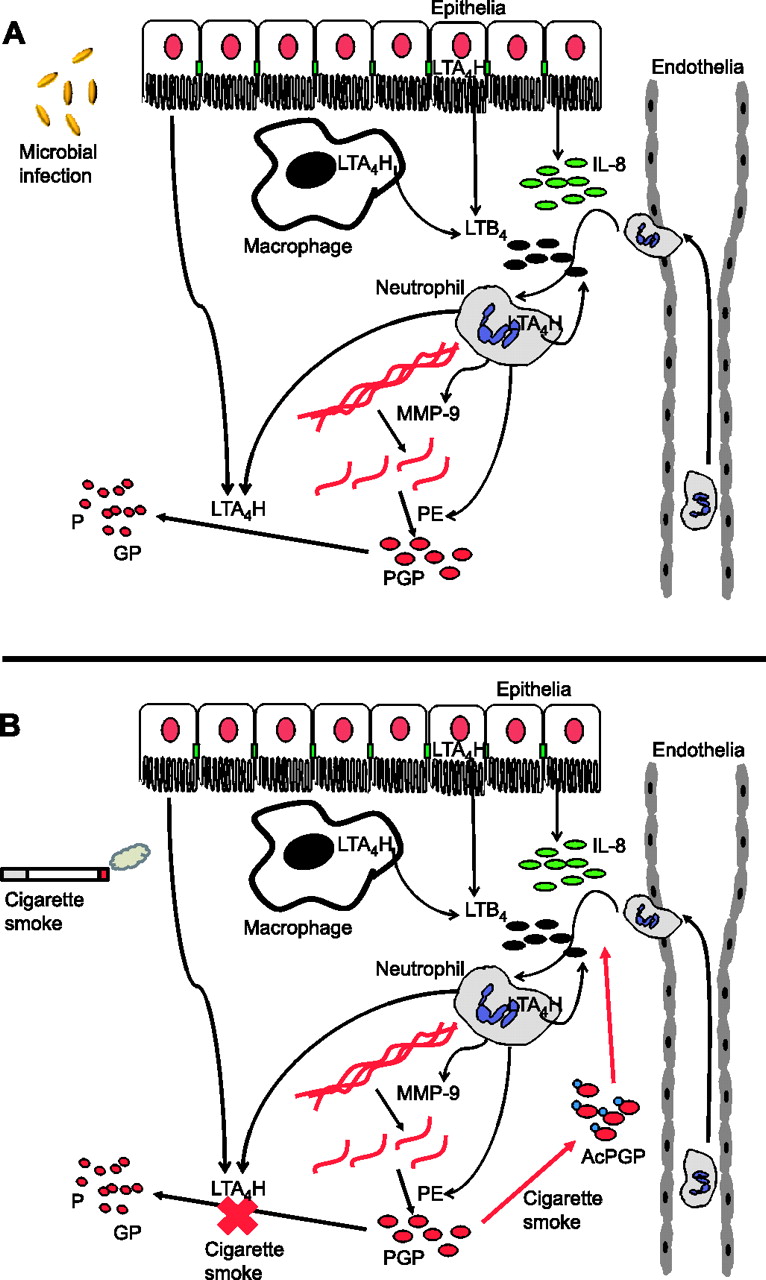
In response to microbial infection or cigarette smoke, lung resident epithelial cells and macrophages release signals, such as interleukin 8 (IL-8) and leukotriene B4 (LTB4), which drive the recruitment and activation of neutrophils. Neutrophils subsequently release enzymes (matrix metalloproteinases (MMPs) and prolyl endopeptidase (PE)) which specifically cleave collagen within the lung to generate the neutrophil chemoattractant peptide Pro-Gly-Pro (PGP). In this manner, neutrophils can themselves promote further neutrophil recruitment leading to persistence and ultimately pathology. (A) To counteract this during microbial infection, leukotriene A4 hydrolase (LTA4H) is released from neutrophils to degrade PGP and resolve neutrophilic inflammation. (B) Cigarette smoke perturbs this LTA4H-mediated anti-inflammatory pathway in two ways: (1) chemically acetylating PGP (AcPGP) and protecting it from degradation by LTA4H while promoting its chemotactic potential; and (2) selective abrogation of LTA4H PGP-degrading activity. As a result, AcPGP/PGP can drive persistent neutrophilic inflammation.
If PGP is normally readily degraded to resolve neutrophilic inflammation, it is rational to question why PGP is present at all in chronic lung diseases with persistent neutrophilia. We demonstrated that cigarette smoke, a major risk factor in the development of COPD, chemically modified PGP by addition of an acetyl group (AcPGP). This modification enhanced the capacity of the peptide to recruit neutrophils and also protected it from degradation by LTA4H. Furthermore, cigarette smoke selectively abrogated the capacity of LTA4H to degrade PGP while having minimal effect on its ability to generate LTB4 (figure 1B). Thus, cigarette smoke seems capable of driving this enzyme, with dual pro- and anti-inflammatory activities, towards a uniquely proinflammatory phenotype whereby LTB4 and PGP can act in tandem to drive the neutrophilic inflammation and pathology observed in COPD. Furthermore, it is intriguing that significant concentrations of PGP/AcPGP are observed in patients with CF, given the defective cystic fibrosis transmembrane conductance regulator-mediated chloride transport in these patients and the fact that chloride ions have been shown to enhance the capacity of LTA4H to degrade PGP. Ultimately, it may be that PGP degradation by LTA4H and acute neutrophilia are the norm and that PGP only persists when this system is perturbed by exogenous stimuli, such as cigarette smoke, or genetic influence.
It is prudent to question the significance of these findings in the context of therapeutic strategies that seek to inhibit LTA4H to reduce LTB4-mediated pathologies. Targeting enzymes crucial to the generation of LTB4 or blocking its receptor binding seem appealing therapeutic targets. Accordingly, there has been a significant pharmaceutical effort to generate LTA4H inhibitors, with Johnson and Johnson and deCODE having developed lead compounds with the latter now in phase II trials. However, these inhibitors seem unlikely to distinguish between the opposing activities of LTA4H and may inadvertently prevent PGP degradation leading to persistent neutrophilia. This is not to say that LTA4H inhibitors should be disregarded out of hand. Indeed, they have shown excellent therapeutic potential in a number of animal models. LTB4 is an extremely potent proinflammatory mediator with many effects on multiple cell types while PGP is far more limited in both its potency and range, so it may be that PGP is the lesser of two evils. The significance of LTA4H in different instances will also be complicated by the availability of enzymes that generate LTA4 and PGP, or a source of acetylation for PGP. It is feasible that the relative importance of each of the LTA4H pathways will be disease- and even patient-specific, but it would be wise to be vigilant to adverse effects of LTA4H inhibitors when testing their efficacy and safety. A series of studies have shown that the opposing activities of LTA4H reside in distinct but overlapping sites within the enzyme, and thus selective modulation of these activities should prove feasible and could offer novel therapeutic avenues to pursue.5
Footnotes
Funding This work was supported by the Wellcome Trust (082727/Z/07/Z).
Competing interests None.
Provenance and peer review Commissioned; internally peer reviewed.