Article Text

Abstract
Background Asthma and chronic obstructive pulmonary disease are airway inflammatory diseases characterised by airflow obstruction. Currently approved bronchodilators such as long-acting β2 adrenoceptor agonists are the mainstay treatments but often fail to relieve symptoms of chronic obstructive pulmonary disease and severe asthma and safety concerns have been raised over long-term use. The aim of the study was to identify the receptor involved in prostaglandin E2 (PGE2)-induced relaxation in guinea pig, murine, monkey, rat and human airways in vitro.
Methods Using an extensive range of pharmacological tools, the relaxant potential of PGE2 and selective agonists for the EP1–4 receptors in the presence and absence of selective antagonists in guinea pig, murine, monkey, rat and human isolated airways was investigated.
Results In agreement with previous studies, it was found that the EP2 receptor mediates PGE2-induced relaxation of guinea pig, murine and monkey trachea and that the EP4 receptor mediates PGE2-induced relaxation of the rat trachea. These data have been confirmed in murine airways from EP2 receptor-deficient mice (Ptger2). In contrast to previous publications, a role for the EP4 receptor in relaxant responses in human airways in vitro was found. Relaxant activity of AH13205 (EP2 agonist) was also demonstrated in guinea pig but not human airway tissue, which may explain its failure in clinical studies.
Conclusion Identification of the receptor mediating PGE2-induced relaxation represents a key step in developing a novel bronchodilator therapy. These data explain the lack of bronchodilator activity observed with selective EP2 receptor agonists in clinical studies.
- Relaxation
- airway smooth muscle
- prostanoids
- prostanoid receptors
- asthma pharmacology
- respiratory muscles
- asthma pharmacology
- respiratory muscles
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Relaxation
- airway smooth muscle
- prostanoids
- prostanoid receptors
- asthma pharmacology
- respiratory muscles
- asthma pharmacology
- respiratory muscles
Key messages
What is the key question?
New bronchodilator therapies are needed, given that currently approved bronchodilators such as long-acting β2 adrenoceptor agonists often fail to relieve symptoms of COPD and severe asthma and safety concerns have been raised over long-term use.
What is the bottom line?
A number of clinical studies have reported beneficial effects of inhaled PGE2 on airway calibre in normal subjects and patients with asthma and COPD, and we have identified a role for the EP4 receptor in these relaxant responses.
Why read on?
Identification of the receptor mediating PGE2-induced relaxation represents a key step in developing a novel bronchodilator therapy.
Introduction
Asthma and chronic obstructive pulmonary disease (COPD) are inflammatory diseases of the airway characterised by airflow limitation. The Global Burden of Disease Study1 has projected that COPD, which ranked sixth as the cause of death in 1990, will become the third leading cause of death worldwide by 2020. Globally, approximately 300 million people have asthma, which is the most common chronic disease in children.2 3
Currently, the majority of patients with inflammatory diseases of the airway are treated with a combination of long-acting β agonists (LABAs) and corticosteroids, but significant issues exist with these therapies, particularly LABAs. While long- and short-acting β agonists help to provide patients with short-term relief from airflow limitation, they do little to treat the underlying pathology. Furthermore, since the introduction of LABAs, there has been increasing concern that their use may be associated with an increased risk of death from asthma.4
A number of clinical studies have reported beneficial effects of inhaled prostaglandin E2 (PGE2) on airway calibre in normal subjects5–8 and in patients with chronic bronchitis and asthma.5 9 10 In addition, PGE2 has anti-inflammatory properties in patients with asthma,9 11 providing an ideal dual therapy for the treatment of these diseases. Despite the benefits of inhaled PGE2, the development of prostanoid agonists for the treatment of airway inflammatory diseases has been hindered as prostanoids induce irritancy of the upper airway resulting in a reflex cough.11 PGE2 can act on a number of receptors, so it was hoped that the beneficial bronchodilator and anti-inflammatory effects could be dissociated from the airway irritancy to provide a novel therapeutic target.
There are currently nine known prostanoid receptors. PGE2 has relatively low affinity for the FP, IP, TP, DP and CRTh2 receptors and acts predominantly via the EP receptors. The EP receptors have been subclassified into EP1, EP2, EP3 and EP4 (encoded by Ptger1–Ptger4 genes),12–14 and distinct signalling pathways initiate diverse and opposing downstream effects in different tissues.13–15 Several investigators have documented bronchodilator activity in vitro and in vivo in a wide range of species including guinea pigs, mice and humans, and it has been suggested that EP2 receptor activation is responsible for PGE2 activity.16–19 However, a selective EP2 receptor agonist (AH13205) with a promising preclinical profile did not appear to have any bronchodilator activity in man.16 Previous studies have been hampered by the lack of selective pharmacological tools. We now have a wide range of tool compounds and, by adopting a pharmacological approach and prostanoid receptor-deficient mice, we provide substantial evidence that, although the EP2 receptor mediates PGE2-induced relaxation in several species, it is in fact the EP4 receptor that mediates relaxation of human airways. This is an important finding which could lead to the development of a new class of bronchodilator therapy.
Methods
Male C57BL/6 mice (18–20 g), Dunkin-Hartley guinea pigs (300–500 g) and Cynomolgus (Cyno) monkey (Macaca fascicularis) tissue were purchased from Harlan (Bicester, Oxon, UK) and male Sprague Dawley rats (250–275 g) were purchased from Charles River (Margate, UK). EP receptor gene-deleted mice were originally provided by Dr Shuh Narumiya, Kyoto University and breeding colonies were maintained at Imperial College, London. Human airway samples (trachea, major bronchus, secondary bronchi) were obtained from donor patients (n=8, 3 male) or recipients (emphysema, n=5, 2 male; cystic fibrosis, n=3, 1 male) for lung transplants performed at the Royal Brompton or Harefield Hospital. Approval was obtained from the Royal Brompton and Harefield ethics committee after receiving the relevant consent from patients and relatives.
The trachea (apart from murine) was opened longitudinally by cutting through the cartilage directly opposite to the smooth muscle layer. Unless stated otherwise, the epithelium was left intact and transverse segments were prepared and sutured in order to be suspended from steel hook transducers in 10 ml baths containing Krebs–Henseleit (KH) solution warmed to 37°C and bubbled with 95% O2/5% CO2. Indomethacin (10 μM), a non-selective COX inhibitor, was present in KH solution in all experiments to prevent the production and release of endogenous prostanoids. Changes in force were measured isometrically using force displacement transducers connected to a data acquisition system as previously described.20
Carbachol (CCh; 1 μM) was used to induce increased tension in tissues over a 30 min period before the addition of any antagonists or their appropriate vehicles which were incubated for a further 30 min. This concentration of CCh was required to produce approximately 80% of the supramaximal response to 1 mM ACh in all species. Cumulative concentration–response curves were then constructed using the relevant drugs and subsequently, at the end of the experiment, the non-specific phosphodiesterase inhibitor papaverine (100 μM) was used to assess the maximum capacity for relaxation of each tissue. For experiments on smaller airways, third to fourth generation bronchi (1–2 mm diameter) were dissected and sutured as described. For these experiments, a higher concentration of CCh (10 μM) was necessary to induce the required level of increased tension.
Full details are given in the online supplement.
Data analysis and statistics
Data are expressed as mean±SEM of n independent observations. Concentration–response curves were analysed by non-linear regression using the PRISM curve-fitting program to produce curves of best fit, from which EC50 values were subsequently derived. Estimates of antagonist affinity were calculated using the equation pKB=log (CR − 1) − log [B] as described by Gaddum,21 where CR is the concentration ratio calculated from the EC50 of agonist in the presence of the antagonist divided by the EC50 of the agonist alone, KB is the equilibrium dissociation constant and [B] is the concentration of antagonist. In the experiments described here, the term pA2 is substituted for pKB as antagonists were used at one concentration only, which precludes assumptions being made about the nature of the antagonism.
Results
Effect of PGE2 on guinea pig tracheal strips
Under basal levels of tension (basal tone), increasing concentrations of PGE2 (0.1 nM–10 μM) resulted in a biphasic response producing a pronounced contraction at lower concentrations (35.4±4.0% of the maximum contraction to ACh, 10 mM) that was later attenuated by increasing the concentration of PGE2 in the bath (figure 1A). This contractile response was completely absent in the presence of the EP1 antagonist GW848687X (1 μM). This was then confirmed using selective EP receptor agonists, with the EP1 selective agonist ONO-D1-004 being the most potent and effective at inducing contraction of the guinea pig trachea (figure 1B).

Involvement of prostanoid EP receptors in prostaglandin E2 (PGE2)-induced contraction and relaxation of guinea pig tracheal strips. (A) Inhibition of PGE2-induced contraction under basal tone by the EP1 antagonist (GW848687X, 1 μM) (PGE2 + the appropriate vehicle (filled circle) or GW848687X in the presence of PGE2 (square), n=4–6). (B) Response to selective EP receptor agonists under basal tone (ONO-D1-004 (filled circle), ONO-AE1-259 (circle), ONO-AE-248 (filled square) and ONO-AE1-329 (square)=EP1–4 respectively, n=4). (C) Inhibition of PGE2-induced relaxation under induced tone by AH6809 (10 μM, EP1/EP2/DP antagonist), (PGE2 + vehicle (filled circle), PGE2 + AH6809 (square), n=4). (D) Response to selective EP receptor agonists under induced tone (ONO-AE1-259 (circle), ONO-AE-248 (filled square) and ONO-AE1-329 (square)=EP2–4 respectively, n=4). Data represent mean±SEM.
When tension was induced in the guinea pig trachea using CCh (1 μM) (induced tone), increasing concentrations of PGE2 (0.1 nM–10 μM) produced substantial relaxation (69.1±7.3% of the maximum relaxation to 100 μM papaverine). Due to the apparent contractile actions of PGE2 via the EP1 receptor, PGE2-induced relaxation in guinea pig trachea was performed in the presence of GW848687X (1 μM). Under these conditions, compared to its vehicle control, addition of AH6809 (10 μM) to the bath before the administration of PGE2 antagonised the relaxation produced (figure 1C), resulting in a rightward shift in the curve (pA2=5.7) commensurate with an action at the EP2 receptor (previous studies report antagonist affinities of approximately 5.9–6.3).22–24 Subsequently, the response to PGE2 was also assessed in the presence of selective antagonists for EP3, EP4, IP and FP receptors (L-826266, 1 μM; GW627368X, 1 μM; RO3244794, 10 μM; and AL-8810, 10 μM, respectively). Compared with their vehicle controls, addition of these antagonists did not produce any effect on PGE2-induced relaxation in guinea pig tracheal strips (figures not shown). When increasing concentrations of each EP receptor agonist (0.1 nM–1 μM) was applied to guinea pig tissue, only the EP2 agonist ONO-AE1-259 induced substantial relaxation (52.8±3.62% of the maximum relaxation to 100 μM papaverine) (figure 1D). These data, alongside the AH6809 data, indicate that the EP2 receptor mediates PGE2-induced relaxation in guinea pig trachea consistent with previous data.16
Effect of PGE2 on murine tracheal segments
In contrast to the guinea pig, the addition of increasing concentrations of PGE2 to mouse tracheal tissue under basal tone failed to produce a contractile response (data not shown). However, when applied to murine tracheal segments under induced tone, PGE2 induced a substantial relaxation response in the tissue (92.6±3.1% of the maximum response to 100 μM papaverine) (figure 2A). This response to PGE2 observed in tissue taken from C57BL/6 wild type mice was then compared with that produced in tissue from prostanoid receptor gene-deficient mice. Of the gene-deleted tissues tested (deficient for EP1, EP2, EP3, EP4, IP, FP, DP1 or TP receptors), only the response of tracheal segments taken from Ptger2 (EP2)-deficient mice differed substantially from tissue taken from wild type equivalents (figure 2C,D). The relaxation response produced by PGE2 in Ptger2−/− tracheal tissue was substantially reduced (24.5±6.6% of the maximum response to 100 μM papaverine), indicating that PGE2-induced relaxation in mouse tracheal tissue is largely dependent on EP2 receptors. Furthermore, AH6809 antagonised PGE2-induced relaxation in mouse tracheal segments under induced tone, producing a rightward shift in the curve (pA2=5.9) commensurate with affinity at the EP2 receptor (figure 2B). Interestingly, there was a small reduction in the relaxation produced by PGE2 in IP receptor-deficient tissues (72.0±5.8% of the maximum response to 100 μM papaverine vs 85.9±1.8% for the wild type control), possibly indicating a minor role for the IP receptor in PGE2-induced relaxation in mouse tracheal segments. The maximum contractile responses to ACh and the maximum relaxation responses to papaverine were not changed in the prostanoid receptor deficient animals.
Involvement of the prostanoid EP receptors in prostaglandin E2 (PGE2)-induced relaxation of murine tracheal segments under induced tone. (A) PGE2-induced relaxation (PGE2 (filled circle), ethanol vehicle control (circle), n=5). (B) Inhibition of PGE2-induced relaxation by AH6809 (10 μM, (EP1/EP2/DP1 receptor antagonist). (PGE2 + vehicle (filled circle), PGE2 + AH6809 (square), n=4). (C) Effect of specific individual EP receptor gene deficiency on PGE2-induced relaxation compared with the respective response elicited in wild-type tissues (C57BL/6 wild type (filled circle), Ptger 1−/− (square), Ptger 2−/− (filled inverted triangle), Ptger 3−/− (circle), n=4). (D) Effect of specific individual EP receptor gene deficiency on PGE2-induced relaxation compared with the respective response elicited in wild-type tissues (129/Ola × C57BL/6 wild type (filled circle), Ptger 4−/− (circle), n=4). Data represent mean±SEM.
Effect of PGE2 on rat tracheal strips
PGE2 (1 nM–10 μM) did not induce a contractile response in rat tracheal strips under basal tone. However, under induced tone, PGE2 produced a concentration-dependent relaxation although smaller in magnitude than in other species (31.9±1.5% of the maximum response to 100 μM papaverine). EP receptor selective agonists (1 nM–10 μM) were evaluated and only the EP4 agonist ONO-AE1-329 caused substantial relaxation (36.7±4.0% of maximum relaxation to 100 μM papaverine) (figure 3A). Furthermore, two structurally distinct EP4 antagonists inhibited PGE2-induced relaxation compared with their vehicle control (0.1% DMSO) (figure 3B). Both GW627368X (1 μM) and ONO-AE3-208 (100 nM) produced a substantial rightward shift in the curve (pA2=6.5±0.1 and 8.9±0.1, respectively) commensurate with an action at the EP4 receptor (previous studies report antagonist affinities of approximately 7 and 8.9 for GW627368X and ONO-AE3-208, respectively25 26). Finally, the effect of removing the epithelium on the response of rat trachea to ONO-AE1-329 was investigated. When the responses of tracheal strips with the epithelium intact were compared with tracheal strips with epithelial cells removed, there was no substantial difference in relaxation to ONO-AE1-329 (epithelium intact: logEC50=−7.5, maximum relaxation=338.3±99.7 mg; epithelium removed: logEC50=−7.6, maximum relaxation=310.5±74.2 mg).

Involvement of the prostanoid EP receptors in prostaglandin E2 (PGE2)-induced relaxation of rat tracheal strips and monkey airway samples under induced tone. (A) Response of rat tracheal strips under induced tone to selective EP receptor agonists (ONO-D1-004 (filled circle), ONO-AE1-259 (circle), ONO-AE-248 (filled square) and ONO-AE1-329 (square)=EP1–4 respectively, n=3). (B) Inhibition of PGE2-induced relaxation of rat tracheal strips by ONO-AE3-208 (100 nM) and GW627368X (1 μM) (PGE2 in the presence of the appropriate vehicle (filled circle), PGE2 in the presence of ONO-AE3-208 (square) or GW62768X (circle), n=4). (C) Response of Cyno monkey airway samples under induced tone to selective EP receptor agonists (ONO-D1-004 (filled circle), ONO-AE1-259 (circle), ONO-AE-248 (filled square) and ONO-AE1-329 (square)=EP1-4, respectively, and PGE2 (filled triangle), n=3). Data represent mean±SEM.
Effect of PGE2 on Cyno monkey tissue
PGE2 produced a concentration-dependent relaxation of induced tone (53.4±5.9% of the maximum relaxation response to 100 μM papaverine) (figure 3C). Of the selective agonists, only the EP2 selective agonist ONO-AE1-259 caused relaxation (36.5±8.0% of the maximum relaxation response to 100 μM papaverine), therefore indicating that, like guinea pig and mouse, PGE2-induced relaxation of Cyno monkey trachea appears to be mediated by the EP2 receptor.
Effect of PGE2 on human airways
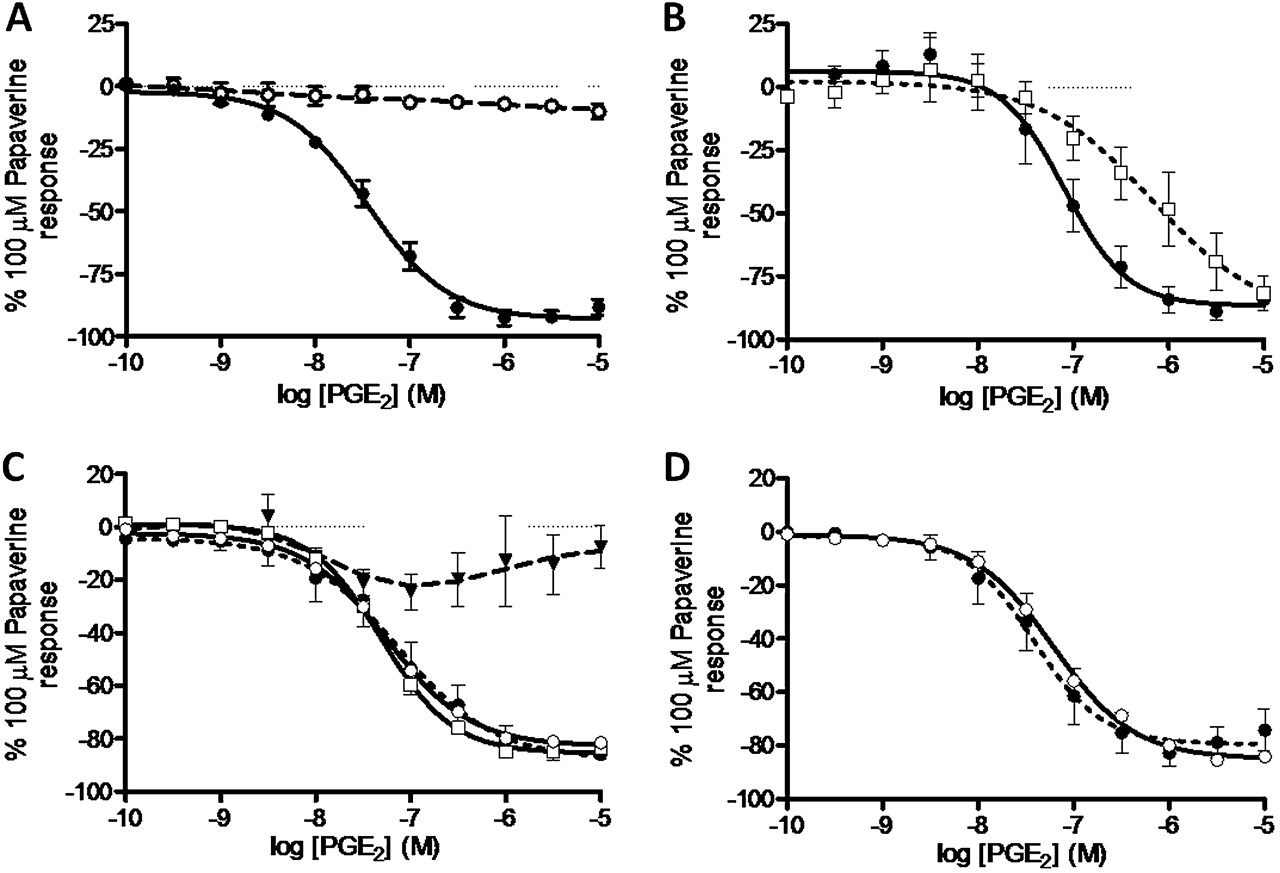
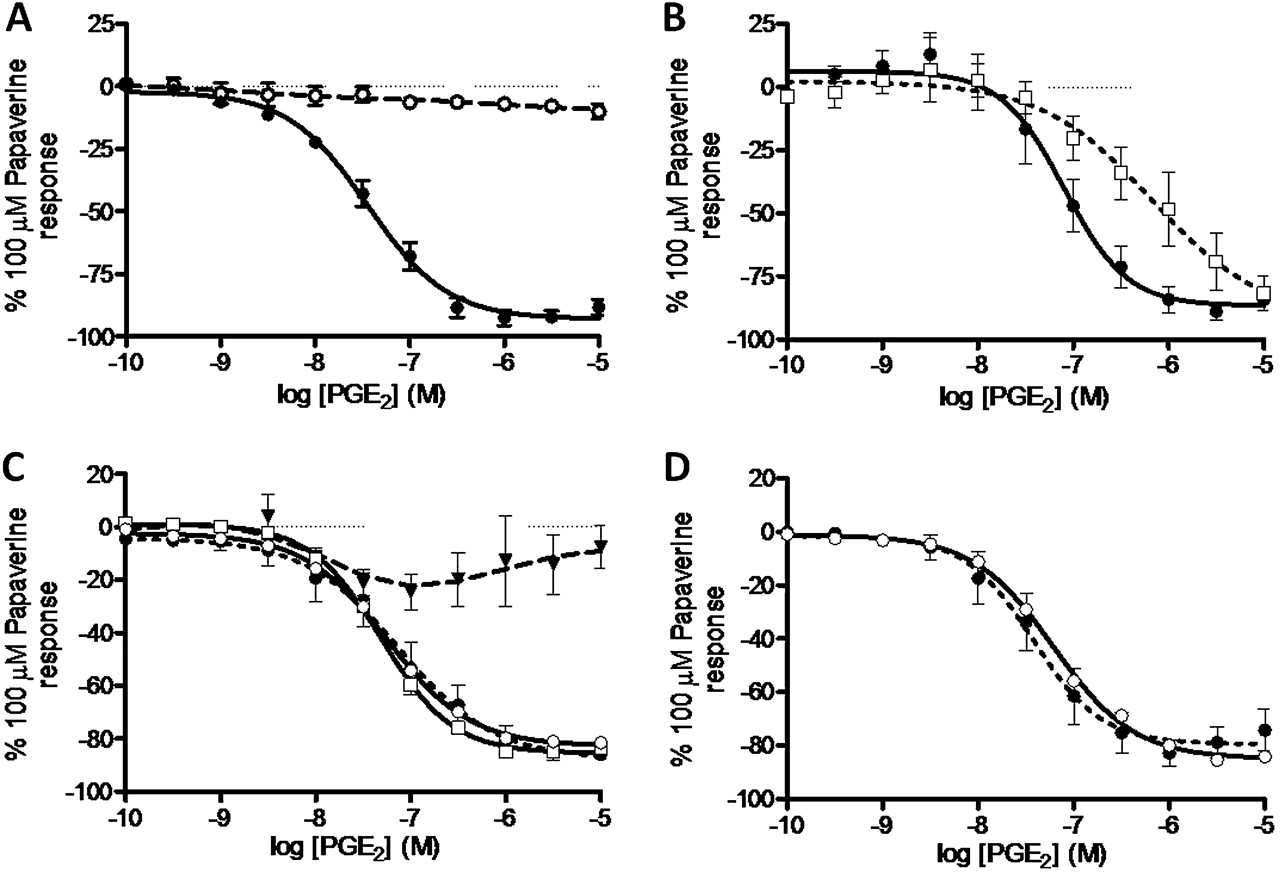
PGE2 (0.1 nM–1 μM) failed to produce a contractile response under conditions of basal tone but produced significant relaxation under conditions of induced tone (62.3% of the maximum relaxation to 100 μM papaverine; data not shown). For comparison, the clinically used LABA formoterol produced 73.8±7.1% of maximum relaxation to papaverine with logEC50=−8.9±0.2, consistent with values in previously published data.27 Of the selective agonists tested, only the EP4 selective agonist ONO-AE1-329 produced a substantial degree of relaxation (76.2±8.6% of maximum relaxation to 100 μM papaverine) (figure 4A). AH6809 failed to antagonise PGE2-induced relaxation in human airway samples (figure 4B). Furthermore, increasing concentrations of two EP2 agonists—AH13205 (10−10–10−5 M) and ONO-AE1-259 (1 nM–10 μM)—failed to produce any significant relaxation (figures 4A and 5B). In experiments on human tissue we used the tool compound with the most optimal profile with regard to antagonist affinity and selectivity (ONO-AE3-208).26 The EP4 antagonist ONO-AE3-208 (100 nM) inhibited both PGE2- and ONO-AE1-329-induced relaxation (pA2=8.0±0.2 and 8.7±0.3, respectively; figure 4C,D). Experiments comparing PGE2 with ONO-AE3-208 in human tissue were conducted in the presence of SQ29548 (TP antagonist, 1 μM) to prevent PGE2-induced contraction via TP receptors. The relaxation response to ONO-AE1-329 was also tested against an alternative non-muscarinic spasmogen. In the presence of 10 μM histamine-induced tone, ONO-AE1-329 still produced substantial relaxation (63.4±19.6% of maximum relaxation to papaverine, logEC50=−7.6±0.5). To ensure the EP4-induced relaxation was not specific to larger airways and not subject to regional differences in the airways, the effect of PGE2, ONO-AE1-259 and ONO-AE1-329 was measured in smaller calibre airways (third to fourth generation bronchi, 1–2 mm diameter). Responses in these airways were similar to those seen in trachea/primary bronchi with PGE2 and ONO-AE1-329 producing relaxation (PGE2: 43.1±5.4% of maximum relaxation to papaverine, logEC50=−7.2 (n=2); ONO-AE1-329: 32.1±5.0% of maximum relaxation to papaverine, logEC50=−7.6±0.1 (n=3)), with ONO-AE1-259 having no substantial effect.

Involvement of EP prostanoid receptors in prostaglandin E2 (PGE2)-induced relaxation of human airway smooth muscle under induced tone. (A) Comparative effects of selective EP receptor agonists (ONO-D1-004 (filled circle), ONO-AE1-259 (circle), ONO-AE-248 (filled square) and ONO-AE1-329 (square)=EP1–4, respectively, n=3–4). (B). PGE2-induced relaxation was not inhibited in the presence of AH6809 (EP1/EP2/DP antagonist, 10 μM) (PGE2 + vehicle (filled circle), PGE2 + AH6809 (circle), n=8). (C) Inhibition of PGE2-induced relaxation by ONO-AE3-208 (EP4 antagonist, 100 nM) in the presence of SQ29548 (TP antagonist, 1 μM) (PGE2 + vehicle and SQ29548 (filled circle), PGE2 + ONO-AE3-208 and SQ29548 (square), n=4). (D) Inhibition of ONO-AE1-329 (EP4 agonist)-induced relaxation by ONO-AE3-208 (EP4 antagonist, 100 nM) (ONO-AE1-329 + vehicle (filled circle), ONO-AE1-329 + ONO-AE3-208 (square), n=4). Data represent mean±SEM.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
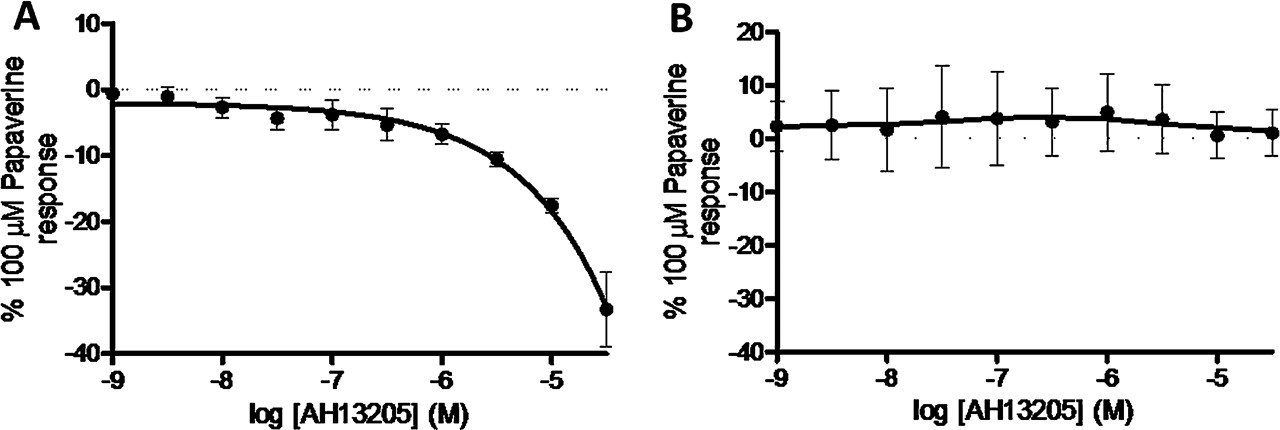
Species differences in the response of airway smooth muscle under induced tone to the EP2 selective agonist AH13205. (A) AH13205-induced relaxation of guinea pig tracheal strips (n=6). (B) Response of human airway smooth muscle to AH13205 (n=3). Data represent mean±SEM.
Finally, in order to confirm that the biological activity of AH13205 was similar to that observed in previous studies, we demonstrated its relaxant activity in guinea pig trachea studies (figure 5A).16 However, this relaxant activity was not parallelled in human airway tissue, which may explain its failure in clinical studies (figure 5B).
Discussion
Inhaled β2 adrenoceptor agonists are currently the gold standard bronchodilators prescribed to patients with respiratory diseases such as asthma and COPD. While long- and short-acting β agonists help to provide patients with short-term relief from airflow limitation, they do little to treat the underlying pathology. Furthermore, since the introduction of LABAs, there has been increasing concern about their safety, particularly when used as monotherapy.4 For these reasons, the search is on for alternative bronchodilator therapies which have an increased safety profile. Identification of a safe therapeutic agent which also possesses anti-inflammatory activity would make for an extremely attractive alternative to LABAs.
PGE2 causes relaxation of animal28 and human17 29 isolated airway smooth muscle. Inhaled PGE2 is a bronchodilator in animals,18 29 30 normal subjects5–8 and in patients with chronic bronchitis and asthma.5 9 10 In addition, PGE2 has anti-inflammatory properties in patients with asthma,9 11 providing an ideal dual therapy for the treatment of these diseases. Despite the benefits of inhaled PGE2, the development of prostanoid agonists for the treatment of airway inflammatory diseases has been hindered as prostanoids induce irritancy of the upper airway resulting in a reflex cough.11
We recently identified the EP3 receptor as being responsible for PGE2-induced airway irritancy (in human, guinea pig and murine in vitro assays) and in a guinea pig cough model, so an opportunity exists to find a therapeutic agent devoid of this sensory irritant side effect if alternative receptors are identified as being responsible for the bronchodilator activity.31 Several investigators have documented bronchodilator activity both in vitro and in vivo in a wide range of species including guinea pigs, mice and humans, and it has been suggested that EP2 receptor activation is responsible for this activity.16–19 However, a selective EP2 receptor agonist (AH13205) with a promising preclinical profile did not appear to have any bronchodilator activity in man.16 Recently, however, improved tools have become available (selective ligands and prostanoid receptor-deficient mice) so, in the light of the issues with current bronchodilators, it seemed pertinent to revisit this area of research.
To investigate which receptor PGE2 acts to cause airway smooth muscle relaxation, a range of selective tools were tested on constricted isolated trachea tissue from guinea pigs, mice, monkeys, rats and humans. In all species PGE2 caused a concentration-dependent relaxation of airway smooth muscle. Experiments using selective ligands on guinea pig, monkey and mouse tissue identified the EP2 receptor as mediating this response, which is consistent with previous studies.16–19 This was further confirmed using mice deficient in individual prostanoid receptors, in that the response to PGE2 present in wild-type animals was substantially diminished in Pgter2−/− mice, again confirming previous data.18 19 30 In contrast, but consistent with previous data, PGE2-induced relaxation of rat isolated tracheal smooth muscle was mediated via activation of the EP4 receptor.32
In contrast, the data from human tissue studies clearly showed that PGE2-induced relaxation is mediated via the EP4 and not the EP2 receptor. This conclusion is based on the in vitro relaxant activity of the selective EP4 receptor agonist (ONO-AE1-329) and the lack of activity of the selective EP2 receptor agonists (ONO-AE1-259, AH13205) in human airways. A role for the EP4 compared with the EP2 receptor was also substantiated by the lack of effect of AH6809 (EP1/2/DP receptor antagonist) and the effectiveness of the EP4 antagonist (ONO-AE3-208) in inhibiting PGE2-induced relaxation of human airway smooth muscle. These data are in contrast to a previous publication describing a role for the EP2 receptor in PGE2-induced relaxation of human airway smooth muscle.17 However, there are some differences between our studies and previous ones which, in the main, can be explained by the lack of selectivity of the tools available at the time the previous studies were carried out. Compared with the study by Norel et al,17 our study has benefited from the use of optimised, more potent and selective agonists (EP2 receptor agonist ONO-AE1-259; EP4 receptor agonist ONO-AE1-329) compared with misoprostol which is a non-selective agonist at EP2–4 receptors33 (table 1). Furthermore, we had the opportunity to use a highly selective EP4 antagonist with greater affinity and selectivity for the receptor than AH23848B, which was used in the previous studies and is a weak antagonist at both the EP4 and TP receptor (pKi 4.9–5.6 and 6.2, respectively), and AH6809, which was used as an EP2 antagonist but also has affinity at the EP1 and DP1 receptors (table 1).33 In the paper by Nials et al,16 the EP2 receptor dependence of the relaxant response (mostly investigated in non-human species) was suggested by the activity of misoprostol, AH13205 and butaprost which all have a relatively weak affinity for the EP2 receptor.16 The relaxant activity of AH13205 in guinea pig trachea described in the study by Nials et al was reproduced in our studies, suggesting that the lack of effect on human airway relaxation was a real phenomenon. The absence of relaxant activity on human airway smooth muscle preparations also provides a likely explanation for the poor efficacy of the EP2 receptor agonist in clinical trials as a bronchodilator in humans.16
Prostanoid EP receptor agonist and antagonist affinities
Interestingly, it would appear that, of all the animal species tested, PGE2-induced relaxation of human airway smooth muscle and the role of the EP4 receptor was most accurately represented in the rat. The responses in non-human primate tissue were not representative of those in human tissue, given the EP2 receptor dependence of the relaxant response to PGE2. This would imply that bioassay of rat tissue would be amenable for medium throughput screening if drug discovery programmes were initiated in this area, thus limiting the use of valuable human airway specimens.
In conclusion, we believe that these data show that the EP4 receptor is responsible for PGE2-induced relaxation of human airway smooth muscle. We recently identified the EP3 receptor as being responsible for PGE2-induced airway irritancy and cough,31 so an opportunity exists to find a therapeutic agent devoid of this sensory irritant side effect liability. However, as PGE2 also enhances plasma leakage in numerous anatomical sites via enhanced blood flow,13 it would be prudent to investigate the receptor responsible as an ideal bronchodilator should not worsen inflammatory exudation or oedema. In summary, EP4 selective compounds could potentially be the next generation of bronchodilators identified since the introduction of β2 adrenoceptors and anticholinergics more than half a century ago.
Acknowledgments
We would like to thank Nicole Dale for her assistance with some of the Cynomolgus (Cyno) monkey tissue experiments.
References
Supplementary materials
Web Only Data thx.2010.158568
Files in this Data Supplement:
Footnotes
Funding The research project and SAM were funded by a project grant from the Medical Research Council (MRC) UK (G0800195). MAB was funded by a grant from the MRC (G0800196). DLC was funded by an MRC project grant Experimental Medicine Grant (G0502019). The human tissue experiments in this study were undertaken with the support of the NIHR Biomedical Research Unit in Advanced Lung Disease at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London and partly funded by the NIHR Biomedical Research Unit funding scheme. JB was funded by a Studentship from a Capacity Building Award in Integrative Mammalian Biology funded by BBSRC, BPS, HEFCE KTN and MRC.
Competing interests None.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the Royal Brompton and Harefield ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.