Article Text

Abstract
Background: In the last decade, Hermansky–Pudlak syndrome (HPS) has arisen as an instructive disorder for cell biologists to study the biogenesis of lysosome related organelles (LROs). Of the eight human HPS subtypes, only subtypes 1 through 5 are well described.
Aim: To characterise extensively the HPS-6 subtype, caused by defects in HPS6, a subunit of the biogenesis of lysosome related organelles complex-2 (BLOC-2).
Methods: Mutation analysis for the HPS6 gene was performed on DNA from our group of unclassified HPS patients. The clinical phenotype of patients with HPS6 mutations was then carefully ascertained, and their cultured dermal melanocytes were employed for cellular immunofluorescence studies.
Results: Molecular studies showed a variety of mutations in the single exon HPS6 gene, including frame shift, missense, and nonsense mutations as well as a ∼20 kb deletion spanning the entire HPS6 genomic region. Cellular studies revealed that the melanogenic proteins tyrosinase and tyrosinase related protein 1 failed to be efficiently delivered to the melanosomes of HPS-6 patients, explaining their hypopigmentation. Clinical studies indicated that HPS-6 patients exhibit oculocutaneous albinism and a bleeding diathesis. Importantly, granulomatous colitis and pulmonary fibrosis, debilitating features present in HPS subtypes 1 and 4, were not detected in our HPS-6 patients.
Conclusion: The HPS-6 subtype resembles other BLOC-2 defective subtypes (that is, HPS-3 and HPS-5) in its molecular, cellular and clinical findings. These findings are not only important for providing a prognosis to newly diagnosed HPS-6 patients, but also for further elucidation of HPS function in the biogenesis of LROs.
Statistics from Altmetric.com
Hermansky–Pudlak syndrome (HPS; MIM 203300) is an autosomal recessive disorder presenting with oculocutaneous albinism, a platelet storage pool deficiency and, in some cases, neutropenia, granulomatous colitis, or a fatal pulmonary fibrosis.1 These clinical features result from defects in the synthesis, processing, or trafficking of lysosome related organelles (LROs), which include melanosomes in melanocytes, delta granules in platelets, lamellar bodies in pulmonary type II cells, and secretory granules in T cells.2 3 To date, eight known human HPS genes have been identified; defects in these genes result in subtypes HPS-1 to HPS-8,3 4 and each human subtype is associated with a murine model.5
The product of the HPS-2 gene, AP3B1, codes for the beta 3A subunit of adaptor complex-3 (AP-3), involved in vesicle formation and protein sorting.6 The exact function of the other HPS gene products remains unknown. All known human HPS proteins interact with each other in biogenesis of lysosome related organelle complexes (BLOCs); HPS1 and HPS4 form BLOC-37 8; HPS3, HPS5 and HPS6 comprise BLOC-29; and HPS7 and HPS8 are members of BLOC-1.10 11 12 Human HPS subtypes involving defects in the same BLOC generally resemble each other clinically.3 4 BLOC-3 deficient patients exhibit a relatively more severe phenotype of hypopigmentation, frequently develop granulomatous colitis, and are the only group who suffer a fatal pulmonary fibrosis.3 4 13 14 Of the BLOC-2 deficient patients, only HPS-3 and HPS-5 have been studied in detail15 16; both have a milder phenotype of variable hypopigmentation and sporadic granulomatous colitis, but pulmonary fibrosis does not occur in HPS-3 or HPS-5 patients. So far, only two BLOC-1 patients have been reported, one HPS-710 and one HPS-8 family11; no detailed clinical features were provided apart from hypopigmentation and a bleeding diathesis.
An accurate diagnosis of the HPS subtype has important prognostic and treatment implications; the neutropenia in HPS-2 can be treated with granulocyte-cell stimulating factor (G-CSF)17 and the pulmonary function in BLOC-3 patients can be addressed in studies of the small molecule pirfinidone.18 Comprehensive genetic analysis of our National Institutes of Health (NIH) cohort of 232 HPS patients over the past decade resulted in detailed clinical and cellular descriptions of the HPS-1,14 19 20 21 HPS-2,6 17 HPS-3,15 19 HPS-4,13 and HPS-516 22 subtypes.
A defect in HPS6, the human homologue of the murine ruby-eye gene, was initially identified in one patient.23 Later we found HPS6 to be defective in an Israeli Bedouin tribe.24 Here we report extensive molecular analysis of HPS6, a single exon gene located on chromosome 10q24.3. Using DNA from our group of unclassified HPS patients, we identified four HPS-6 patients and seven novel HPS6 mutations. We ascertained the clinical phenotype of HPS-6 disease (MIM607522), and we employed HPS-6 patients’ melanocytes for cellular studies.
Patients and methods
Patients and cells
All patients were enrolled in a protocol approved by the National Human Genome Research Institute (NHGRI) institutional review board to evaluate the clinical and molecular aspects of HPS. Written informed consent was obtained from the patient or the patient’s parent. The diagnosis of HPS was based on decreased visual acuity, nystagmus, hypopigmentation, and the absence of platelet dense granules on whole mount electron microscopy.1 Primary cultures of epidermal melanocytes and fibroblasts were obtained from a forearm skin biopsy and cultured as previously described.15 22
Electron microscopy of platelet dense bodies
Platelet-rich plasma, prepared from fresh citrated blood, was placed on copper grids and treated as described.1 15 The grids were air dried and examined using a Philips model 301 electron microscope.
Molecular analysis
Genomic DNA and RNA were extracted from cultured fibroblasts or peripheral leucocytes as described.15 Primers were designed to amplify the coding region, the 3’UTR and a part of the 5’UTR encoded by the single HPS6 exon in nine overlapping fragments from genomic DNA (primer sequences available on request). Standard polymerase chain reaction (PCR) amplification procedures were employed. All PCR amplified products were directly sequenced using standard protocols. To verify allelic expression of the mutations in patient HPS115, a PCR fragment spanning both mutations was cloned into the TOPO TA Cloning Kit per the manufacturer’s instructions (Invitrogen, Carlsbad, California, USA). Individual clones were purified using the QIAprep Spin Miniprep Kit (Qiagen, Valencia, California, USA) and subjected to sequence analysis. To assess carrier frequency of the c.815C>T (p.T272I) variant, 197 Caucasian DNA samples (394 chromosomes) were acquired from the HD200CAU panel from the Coriell Institute for Medical Research (Camden, New Jersey, USA) and tested by BglII restriction enzyme analysis. For this assay, DNA was PCR amplified with forward primer 5′- CTGGGTTGCTGTCCCCCAGGGAGCCACTGGCTGTACAGA-3′ and reverse primer 5′-CATGTCCAGCAGTTCCAATG-3′, yielding a 236 bp fragment. The forward primer was modified so that in the amplified mutated allele (T-nucleotide) a BglII restriction site was created (yielding 199 bp and 37 bp fragments after BglII cutting), while the normal allele (C-nucleotide) would remain uncut by BglII (supplemental fig S1).
Multiple tissue northern blot analysis
A human multiple tissue northern blot was obtained from Clontech (Palo Alto, California, USA) and hybridised as described.15 The HPS6 probe was prepared by PCR amplifying gDNA with primers 5′-GAAGCCCCACAGGGTGTTGAGTTG-3′ (nt 1250–1273) and 5′-GTGTCAGTGACACAGAGGAACTGT-3′ (nt 2478–2501) (GenBank NM_024747). In addition, the filter was probed with a β-actin probe (Clontech).
Copy number real-time quantitative PCR
TaqMan primers and probes (supplemental table S1) were ordered as pre-manufactured assays on demand (HPS6 assays Hs00544576_s1 and Hs00949672_s1) or custom designed for specific target sequences in the HPS6 genomic region using the ABI Assay-by-Design service (Applied Biosystems, Foster City, California, USA). Genomic DNA of patients HPS51 and HPS107 was PCR amplified for the target sequences and for an endogenous control gene, RNaseP, on an ABI PRISM 7900 HT Sequence Detection System, following the manufacturer’s recommendations (Applied Biosystems). Each PCR amplification was performed in triplicate and each experiment was repeated three times. The comparative Ct method was used to determine the target gene copy number in each sample.25 The primer pair for amplification over the breakpoints of the deleted area in DNA of patient HPS107 were: P1, 5′-GGATGCTTGCCATGTTGCCC-3′ and P2, 5′-GGATCCCGCCATGGTGTCCACC-3′, yielding a fragment of 298 bp that was directly sequenced (fig 1F).
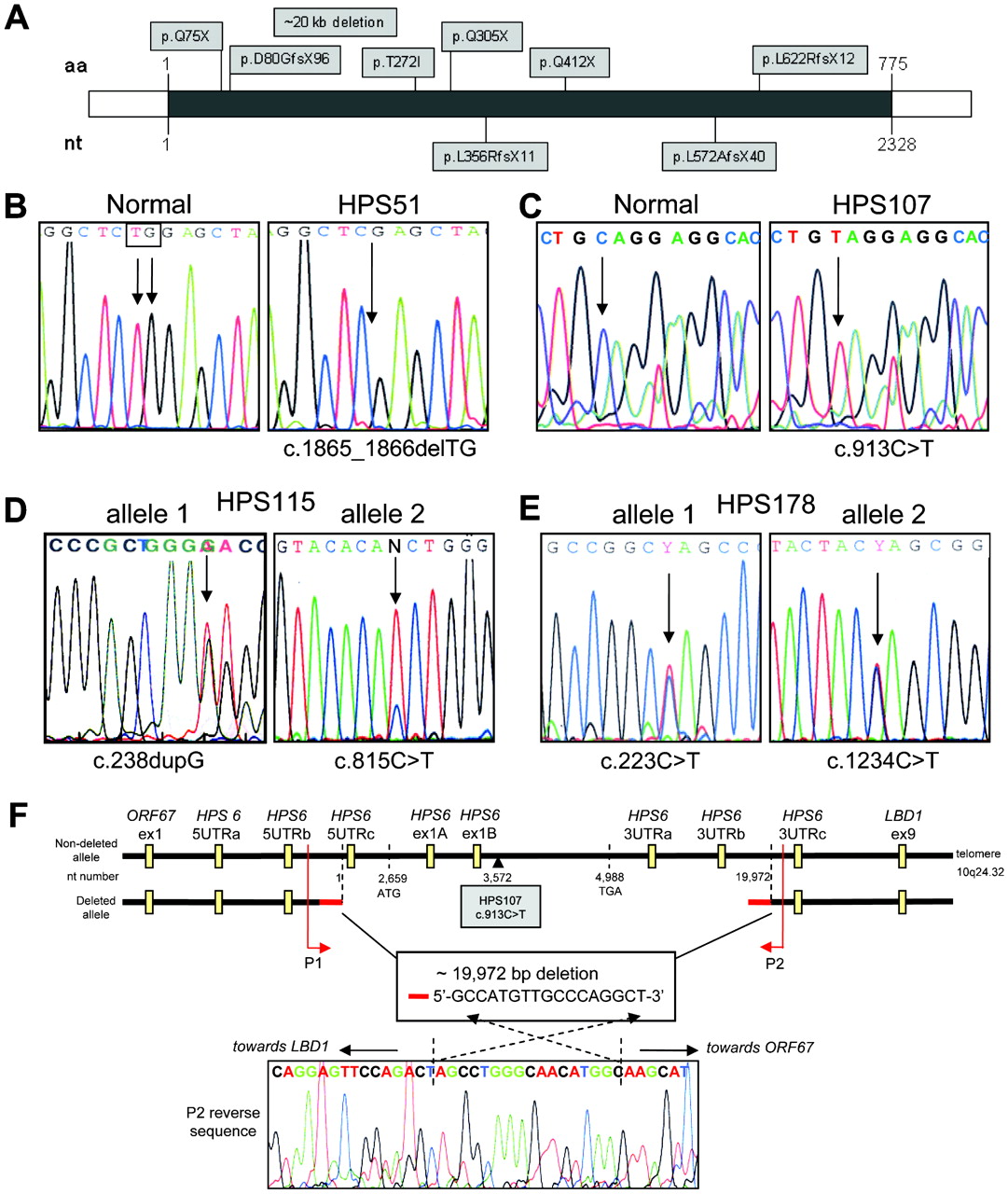
Molecular analysis of HPS-6 patients. (A) All identified mutations in HPS6 to date, indicated by amino acid change. Mutations denoted above the HPS6 gene (grey line) are novel mutations described in this paper. Mutations below the HPS6 gene were previously reported. aa, amino acid; nt, nucleotide. (B) Patient HPS51 was homozygous for c.1865_1866delTG [p.L622RfsX12]. (C) Patient HPS107 was hemizygous for c.913C>T [p.Q305X]. (D) Patient HPS115 was compound heterozygous for a frameshift c.238dupG [p.D80GfsX96] (left panel) and a missense mutation c.815C>T [p.T272I] (right panel). (E) Patient HPS178 was compound heterozygous for two nonsense mutations: c.223C>T [p.Q75X] (left panel) and c.1234C>T [p.Q412X] (right panel). (F) Schematic of the 19972 bp deletion in patient HPS107 (not to scale). Deletion breakpoints, size, overlapping sequence and positions of the Taqman copy number assays are indicated. Primers P1 and P2 were used to sequence across the deletion; the reverse sequence acquired with the P2 primer, including the overlapping 17 bp sequence, is shown.
Immunocytochemistry
For steady state immunofluorescence (endogenous protein distribution), control and patients’ melanocytes were grown overnight on glass chamber slides and fixed in 4% paraformaldehyde. The slides were blocked in PBS containing 0.1% saponin, 100 μM glycine, 0.1% BSA and 2% donkey serum, and then incubated with the following antibodies: mouse monoclonal MEL-5 recognizing TYRP1 (1:200 dilution; Signet Laboratories, Dedham, Massachusetts, USA), HMB45 detecting PMEL17 (1:200 dilution; Lab Vision, Fremont, California, USA), and T311 detecting TYR (1:200; Santa Cruz Biotechnology, Santa Cruz, California, USA). The cells were subsequently washed and incubated with donkey anti-mouse antibodies conjugated to ALEXA-488 (Invitrogen), and nuclei were stained with TO-PRO-3 (Invitrogen). The cells were then rinsed and mounted in VectaShield (Vector Laboratories, Burlingame, California, USA). For TYRP1 internalisation studies, melanocytes were grown on glass slides and incubated for 30 min at 37°C in the presence of MEL-5 antibodies (1:200) diluted in DMEM containing 0.1% (wt/vol) BSA, and 25 mM HEPES buffer, pH 7.4. Subsequently, cells were washed in ice cold PBS, fixed in 4% paraformaldehyde, and incubated with secondary donkey anti-mouse ALEXA-488 antibodies (Invitrogen) and mounted as described above.
All slides were imaged with a Zeiss LSM510 META confocal laser scanning microscope using 20X/.08 or 63X/1.4 Plan-Apochromat objectives (Carl Zeiss, Microimaging Inc, Germany). All images are collapsed stacks of confocal Z sections. Fluorescence intensity plots were created using Zeiss LSM software (Carl Zeiss, Microimaging Inc).
Results
Mutation analysis
Genomic DNA of our group of approximately 30 unclassified HPS patients was screened for mutations in HPS6 by PCR amplification followed by direct sequencing. We identified HPS6 mutations in four patients: HPS51, HPS107, HPS115, and HPS178 (fig 1, table 1).
HPS6 mutations and patients’ ancestries
Patient HPS51 displayed homozygosity for a novel two-base deletion, c.1865_1866delTG [p.L622RfsX12], resulting in a frameshift leading to a premature termination codon at amino acid position 633 with loss of nearly 20% of the HPS6 protein at the 3′ end (fig 1B). Patient HPS107 appeared homozygous for a novel nonsense mutation, c.913C>T [p.Q305X] (fig 1C). Patient HPS115 was compound heterozygous for a frameshift c.238dupG [p.D80GfsX96] and a missense mutation c.815C>T [p.T272I] (fig 1D). Amplification, subcloning and sequencing the HPS6 region that included both mutations confirmed that the mutations were on separate alleles. The c.815C>T (p.T272I) missense mutation is not one of the two reported SNPs in HPS6; c.516G>A [p.G172G; rs3737243] and c.698T>G [p.L233R; rs36078476] (rs numbers refer to dbSNP database: http://www.ncbi.nlm.nih.gov/projects/SNP/, Genome Build 36.3 (accessed 15 September 2009). In addition, we tested 197 Caucasian DNA samples (394 chromosomes) from Coriell panel HD200CAU by BglII restriction enzyme analysis (supplemental fig S1) and we sequenced at least 90 Caucasian HPS patients’ chromosomes for HPS6 mutations; none of these 484 tested chromosomes carried the c.815C>T variant. Furthermore, the threonine at position 272 and its surrounding amino acids (VHTWAP) are conserved in other species (chimpanzee, horse, dog, pig, cow, rat, mouse, chicken), suggesting a functionally important amino acid.
Patient HPS178 was compound heterozygous for two nonsense mutations, c.223C>T [p.Q75X] and c.1234C>T [p.Q412X] (fig 1E).
Both patients HPS51 and HPS107 appeared to be homozygous for a rare mutation, yet their families reported no consanguinity. Therefore, we investigated hemizygosity in these patients by copy number real-time PCR on genomic DNA, initially with two HPS6 Taqman assays located in HPS6 (assays HPS6ex1A and HPS6ex1B in fig 1F, supplemental table S1). Patient HPS51 had two allelic HPS6 copies for each assay, similar to control DNA. However, patient HPS107 had only one allelic copy for each HPS6 assay, and two allelic copies of the control gene (RnaseP), indicating hemizygosity in the HPS6 region. To determine the dimension of this 10q24 deletion in patient HPS107, we performed copy number real-time PCR using a variety of Taqman primer probe assays on either side of the deleted area (fig 1F, supplemental table S1). Two allelic copies were found for assays targeting the upstream gene ORF67 (assay ORF67ex1) and the downstream gene LBD1 (assay LBD1ex9). Subsequent assays targeting the upstream (5’UTR assays) and downstream (3’UTR assays) HPS6 regions narrowed the deleted area to ∼20 kb located between HPS6 5UTRb and HPS6 3UTRc (fig 1F). We then tested combinations of primers on either side of the deletion to amplify across the deletion. Primers P1 and P2 yielded a 298 bp PCR fragment, and sequencing revealed the exact deletion breakpoints (fig 1F). The deletion covers 19 972 bp of genomic DNA, spanning the entire HPS6 coding region, starting 2659 bp upstream of the HPS6 start codon (ATG) and ending 14 984 bp downstream of the HPS6 termination codon (TGA). The breakpoints do not occur in a known genomic repeat area, but both breakpoint areas contain an identical 17 bp sequence: GCCATGTTGCCCAGGCT (fig 1F). This suggests that the deletion is a “simple deletion”, meditated by a “symmetric element” mechanism involving short repetitive sequences present in the same direction on one allele.26
For patient HPS51 we sequenced six single nucleotide polymorphic sites (SNPs) nearby on chromosome 10q24. All SNPs tested showed homozygosity, so uniparental disomy could not be excluded, but not further pursued since DNA from the patient’s parents was not available.
HPS6 gene organisation
HPS6 consists of one exon, located on chromosome 10q24.3. The open reading frame is predicted to code for a 775 amino acid, 83 kDa protein of unknown function. The protein is highly homologous to its mammalian orthologs, but lacks homology to any other protein. No functional domains, leader sequence, or N-glycosylation sites are predicted. A multiple-tissue northern blot demonstrated that HPS6 was expressed in all tissues tested, displaying a transcript size of approximately 2.6 kb, and no alternatively spliced transcripts were present (fig 2A). This ubiquitous expression is similar to that of other HPS genes15 16 and was confirmed by a tissue microarray expression database (Stanford Microarray Database http://smd.stanford.edu/ (accessed 15 September 2009)). Our attempts to create antibodies to the human HPS6 protein were unsuccessful, limiting further HPS6 protein analysis.

HPS6 mRNA expression. A multiple tissue northern blot hybridised with a [32P]dCTP labelled HPS6 probe demonstrating a ∼2.6 kb HPS6 mRNA transcripts in all tissues tested (upper panel). The blot was subsequently hybridised with a [32P]dCTP labelled β-actin probe to control for RNA loading (lower panel).
Cellular studies
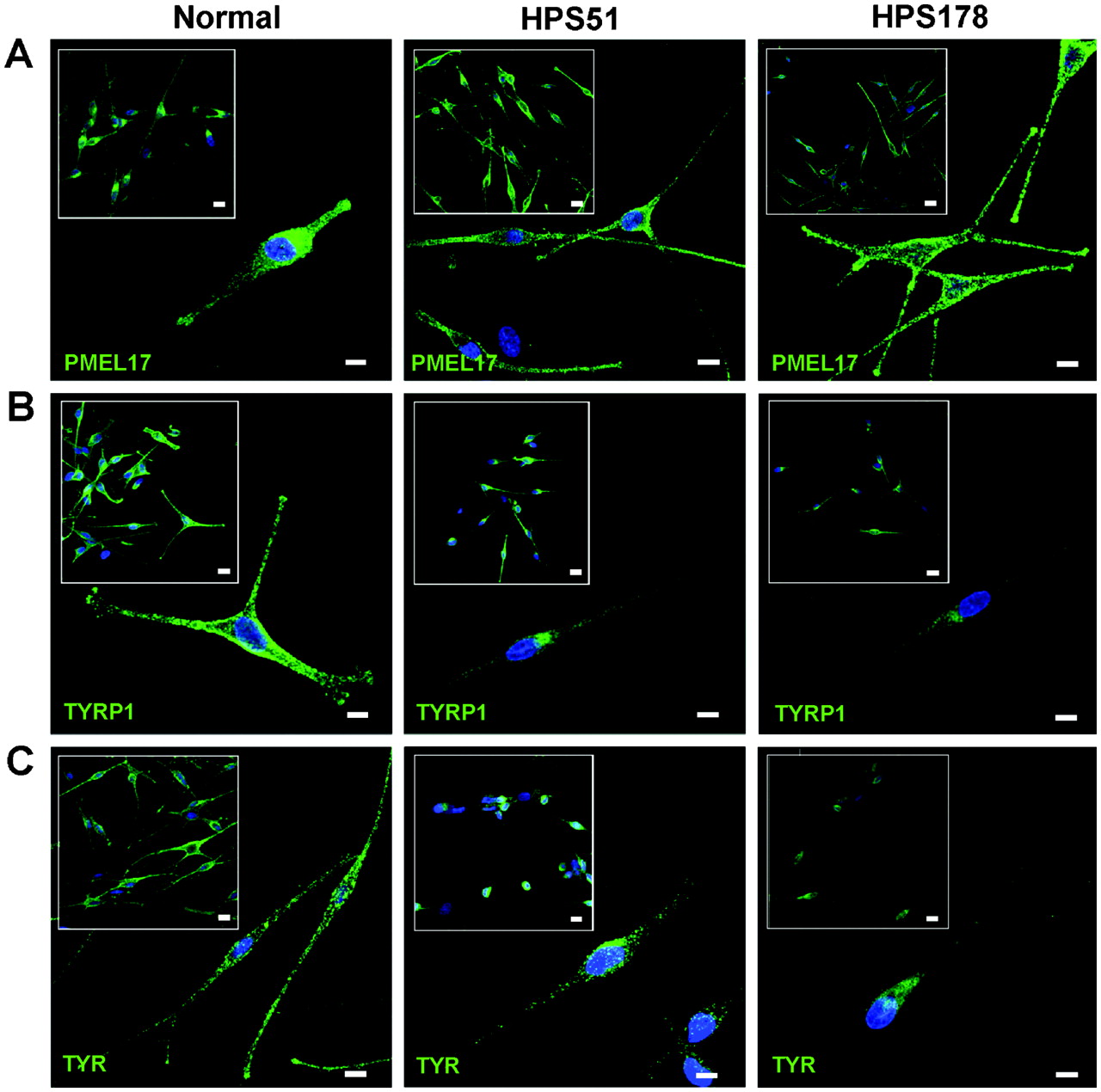
Melanocytes are an ideal cell type to examine the biogenesis of lysosome related organelles.2 3 22 27 To evaluate the HPS-6 cellular phenotype, distributions of melanosomal proteins PMEL17, TYRP1, and TYR were assessed for patients HPS51 and HPS178 (fig 3). These patients’ melanocytes showed a dispersed cytoplasmic distribution of PMEL17, with accumulation in the dendritic tips, similar to the pattern in normal melanocytes (fig 3A). Normal melanocytes also showed a dispersed cellular pattern for TYRP1 and TYR, with accumulation in the dendritic tips (fig 3B,C left panels). However, TYRP1 and TYR distribution in the HPS-6 patients’ melanocytes remained perinuclear, with decreased staining in the cell periphery and little or no accumulation in the dendritic tips (fig 3B,C, right panels). These aberrant patterns were similar to previously reported distribution of TYRP1 and TYR in BLOC-2 deficient human and mouse melanocytes,22 28 29 following the hypothesis that PMEL17 is a resident protein of stage I and II melanosomes, where it serves as the structural foundation of fibrils upon which melanin can be deposited in later stage melanosomes.2 30 Stage I and II melanosomes mature into stage III melanosomes by acquiring other melanogenic proteins and enzymes such as TYRP1 and TYR, which are crucial for melanin production, eventually resulting in a melanin laden stage IV melanosome.2 27 30 Endosomal transport of TYRP1 and TYR from the trans-Golgi complex into a sorting endosome is thought to be mediated by BLOC-3, while BLOC-2 is thought to play a role in further sorting and transport from the sorting endosome to stage II and III melanosomes.2 27 29 Previous studies of BLOC-2 human (HPS5 deficient22) and mouse (cocoa, Hps3 deficient28) melanocytes showed enhanced flux of TYRP1 protein through the plasma membrane and decreased steady state TYRP1 levels due to lysosomal degradation of mis-trafficked TYRP1. Our studies of HPS-6 melanocytes yielded similar results; steady state TYRP1 levels were decreased in HPS51 and HPS178 melanocytes (fig 3B), and TYRP1 internalisation experiments revealed enhanced trafficking of TYRP1 through the plasma membrane leading to increased internalisation of TYRP1 antibodies in HPS-6 melanocytes (fig 4). All images in fig 3 and 4 are representatives of three independent experiments.
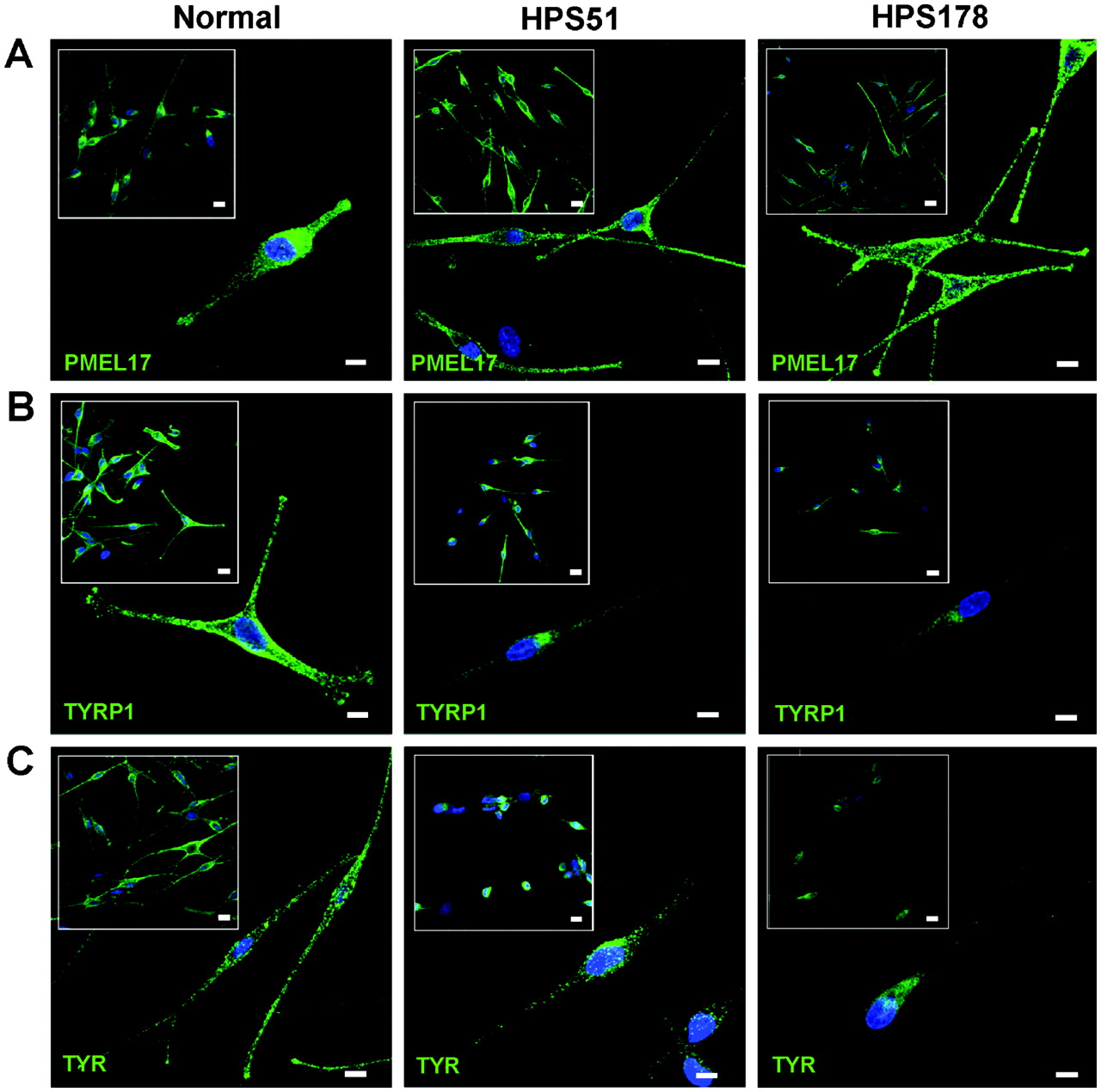
Steady state distribution of melanogenic markers PMEL17, TYRP1, and TYR in normal and HPS-6 patients’ melanocytes (HPS51 and HPS178). (A) PMEL17 (HMB45, green) is normally distributed throughout the perinuclear region and accumulates in the tips in HPS-6 melanocytes. (B) TYRP1 (MEL5, green) is concentrated in the perinuclear region and fails to extend into the dendrites and does not accumulate in the tips of HPS-6 melanocytes. Intensity of TYRP1 signals in HPS-6 cells is decreased due to lysosomal degradation of mis-trafficked TYRP1.22 28 (C) TYR (T311, green) is concentrated in the perinuclear region and does not extend into the dendrites and tips of HPS-6 melanocytes. All cell nuclei are stained with TO-PRO-3 (blue). Images represent collapsed stacks of confocal z-sections. Size bars = 10 μm; size bars in insets = 20 μm.

TYRP1 antibody internalisation studies on HPS-6 melanocytes. (A) Trafficking of TRYP1 (green) via the cell membrane is markedly increased in HPS-6 melanocytes (HPS51 and HPS178) compared to normal melanocytes (left panel), as illustrated by TYRP1 antibody incubation for 30 min in unpermeabilised cells. Cell nuclei are stained with TO-PRO-3 (blue). Images represent collapsed stacks of confocal z-sections. Size bars = 20 μm. (B) Fluorescence intensity plots of the images in (A) (green channel only).
Clinical findings
Patient HPS51
Patient HPS51 is a 36-year-old woman. At 5 months, an eye consultant found horizontal nystagmus and diagnosed partial albinism. The diagnosis of HPS was made at age 26 based upon eye findings and bleeding complications, and was confirmed by the finding of absent platelet dense bodies (fig 5C). The patient has a history of multiple abdominal surgeries for hernia, imperforate anus and gluteal flap repairs. Four units of packed red cells and several platelet transfusions were administered before these procedures. The patient bruises easily and has a bleeding time of 25.5 min. She has had four miscarriages, endometriosis, heavy menses, frequent upper respiratory and urinary tract infections, urinary and rectal incontinence with neurogenic bladder dysfunction, frequent migraines and hearing loss. On admission, the height was 161 cm, weight 92.5 kg, blood pressure 121/78 mm Hg. Hair colour was light brown. Visual acuity was 20/100 in the right eye and 20/125 in the left. Iris transillumination was present (fig 5A), foveal reflex was absent, and the fundus was pale (fig 5B). Whole mount electron microscopy of the platelets showed absent dense bodies (fig 5C). The forced vital capacity (FVC) was 94% of predicted, forced expiratory volume in 1 s (FEV1) was 104% of predicted, and the total lung capacity for carbon monoxide (DLCO) was 90% of predicted. The creatinine clearance was 129 ml·min−1·1.73 m−2 (normal 90–125). A chest radiogram was normal with no evidence of pulmonary fibrosis or interstitial lung disease. The serum cholesterol was 188 mg/dl (normal 100–200) and the triglycerides were 88 mg/dl (normal <150).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Clinical features of HPS-6 patients. Iris transillumination is caused by light that is transmitted back through the iris when light is shone through the pupil. A normal iris remains dark (left panel). All four HPS-6 patients show light transmitted back as a result of reduced iris pigmentation. (B) Contrasting with the normal, uniform pigmentation (left panel), each HPS-6 patient has variable degrees of hypopigmentation of the retina. (C) Whole mount electron micrographs of platelets (×10 000–13 000). Normal platelets contain several delta granules per platelet (left panel, arrows), while HPS-6 platelets lack delta granules.
Patient HPS107
Patient HPS107 is a 22-year-old man. Nystagmus was present at birth and the diagnosis of albinism was made at 3 months. As a toddler, the patient experienced prolonged bleeding and intermittent epistaxis; no other significant bleeding occurred. The diagnosis of HPS was made at age 16 based upon absence of platelet dense bodies (fig 5C). On admission, the height was 179 cm, weight 62 kg, and blood pressure 136/71 mm Hg. The hair was brown, visual acuity was 20/80 in the right eye and 20/63 in the left. There was transillumination of the irides (fig 5A), absence of foveal reflex, and hypopigmentation of the fundi (fig 5B). The FVC was 104% of predicted, FEV1 82% of predicted, and the DLCO 77% of predicted. The creatinine clearance was 138 ml·min−1·1.73 m−2, the serum cholesterol was 143 mg/dl, and triglycerides 306 mg/d. The bleeding time was 9 min. Chest computed tomography (CT0 scan showed two small, non-specific nodular densities, with no focal infiltrates.
Patient HPS115
Patient HPS115 is a 13-year-old girl. At 2 months of age she was noted to have mild horizontal nystagmus and was diagnosed with tyrosinase positive oculocutaneous albinism. Her hair, eyebrows and eyelashes were nearly white during the first year of life, but she developed some pigment later. She had strabismus surgery on both eyes at 14 months without major bleeding. After she began walking at 19 months, she was noted to bruise easily and have very slow nail growth. The patient has global developmental delay. At 14 months she was 6 months behind in both motor function and speech/language. She started using single words at 2–2.5 years of age and progressively acquired function over the next few years. At 4 years, the bleeding time was >20 min, with normal von Willebrand factor, prothrombin time (PT) and partial thromboplastin time (PTT) tests. In vitro platelet aggregation responses to collagen, ADP and arachidonic acid were all abnormal, prompting the diagnosis of platelet storage pool deficiency and HPS. Her platelets demonstrated absence of dense bodies (fig 5C), confirming the HPS diagnosis. She had no epistaxis or any other major bleeding events. At age 8, intravenous desmopressin (DDAVP) and oral aminocaproic acid (Amicar) were used before dental work, which was performed without complications. On admission, the height was 137 cm (75%), weight 42.5 kg (97%), and blood pressure 107/52 mm Hg. Visual acuity was 20/200 bilaterally. The serum cholesterol was 141 mg/dl and the triglycerides 54 mg/dl.
Patient HPS178
Patient HPS178 is a 52-year-old man. His two sisters have HPS, with symptomatic bleeding; one sister also has von Willebrand factor deficiency. The patient had rotatory nystagmus at birth and bruising in childhood. The diagnosis of HPS was made at age 44 based upon the combination of gastrointestinal symptoms, nystagmus and oculocutaneous albinism. A pre-hernia repair evaluation at age 47 revealed a haemoglobin of 9 g/dl (normal 12.7–16.7). Iron and vitamin B12 deficiency were present. The patient had a history of oesophageal dysmotility, hiatal hernia and other gastrointestinal complications including gastro-oesophageal reflux disease, dysphagia, and bloating. On admission, the blood pressure was 159/59 mm Hg. Visual acuity was 20/160 in the right eye and 20/125 in the left. The hair was light brown and the eyes dark brown. His irides showed transillumination (fig 5A). The FVC was 105% of predicted, FEV1 112% of predicted, and the DLCO 105% of predicted. The creatinine clearance was 130 ml·min−1·1.73 m−2, the serum cholesterol 96 mg/dl, and triglycerides 79 mg/dl. Chronic inflammation due to recurrent gastrointestinal complications and an elevated C reactive protein suggested that his anaemia was chronic; it was not considered to be due to vitamin or iron deficiencies. Chest, abdomen and pelvis CT scans showed a calcified granuloma and a few non-specific, sub-centimetre irregular focal densities in the lung. No evidence of interstitial lung disease was detected.
Discussion
Careful evaluation of our group of HPS-6 patients revealed that they appear to have clinical features similar to those of other BLOC-2 deficient patients—that is, patients with HPS-315 and HPS-516—including variable hypopigmentation and bleeding diathesis. Importantly, HPS-6 patients showed no signs of granulomatous colitis or pulmonary fibrosis, which are common features in BLOC-3 deficient patients with HPS-1 and HPS-4.1 13 14 20 21 Specifically, two patients (HPS107 and HPS115) were of an age at which lung disease typically develops in BLOC-3 deficient patients. It is therefore imperative that adults with HPS-6 continue to be followed for the development of restrictive lung disease in order to determine whether pulmonary involvement occurs in this subtype and to anticipate treatment.18
Molecular findings indicated a variety of HPS6 mutations scattered throughout the gene (fig 1A, table 1). All mutations were nonsense or frame-shift, likely resulting in aberrant protein translation. However, one missense mutation, p.T272I, was located in a conserved HPS6 region, possibly indicating significance for HPS6 function. Furthermore, p.T272I variation was not identified in 484 Caucasian chromosomes. Molecular studies on DNA of patient HPS107 demonstrated the importance of considering hemizygosity when a DNA alteration appears to be homozygous. Confirmation of hemizygosity can eliminate concerns about non-paternity and can help explain other clinical features if the deleted area includes other genes.
Cellular studies performed on HPS-6 melanocytes indicated aberrant distribution patterns of the melanogenic proteins TYRP1 and TYR, as well as increased trafficking of TYRP1 through the plasma membrane (figs 3 and 4), similar as those described for other BLOC-2 deficient (HPS3 and HPS5) melanocytes. These findings confirmed that the BLOC-2 subunits HPS3, HPS5, and HPS6 act in the same pathway of LRO biogenesis. Since increased plasma membrane trafficking of TYRP1 does not occur in BLOC-3 deficient cells,28 immunofluorescent microscopy on HPS patients’ melanocytes may assist in diagnosing BLOC-2 deficiency, and focus genetic analyses on the HPS3, HPS5, and HPS6 genes. These cellular studies again denote the value of cultured melanocytes for research of LROs2 3 27 and we encourage that, when possible, a melanocyte culture is grown from skin biopsies of patients with LRO disorders. These cells will be invaluable sources for future investigations into the biogenesis of LROs.
Acknowledgments
Isa Bernardini, Roxanne Fischer, and David Claassen provided excellent technical assistance.
REFERENCES
Supplementary materials
Web Only Data 46/12/803
Files in this Data Supplement:
Footnotes
Additional figure and table are published online only at http://jmg.bmj.com/content/vol46/issue12
Funding This work was supported by the Intramural Research programs of the National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland, USA.
Competing interests None.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.