Article Text

Abstract
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive and ultimately fatal disorder for which there is no cure. While the disease is by definition idiopathic, accumulating evidence, including familial aggregation of cases and the occurrence of pulmonary fibrosis in the context of a number of rare genetic disorders, indicates that genetic factors contribute significantly to the pathogenesis of IPF. Several disease-associated genetic variants, both rare and common, have been identified in familial and sporadic IPF. While the full clinical implications of these genetic associations remain to be elucidated, observational studies suggest that genotype influences the development of the disease and its outcome. Available data indicate that genetics has the potential to identify individuals at risk of IPF, classify patients more precisely, clarify the key pathways involved in disease pathogenesis and eventually develop more effective targeted therapies. Considerable research is required before a comprehensive disease fingerprint of IPF can be delivered. Nevertheless, the application of rapidly evolving molecular biology and genomic technologies combined with appropriate bioinformatic methodology offers an unprecedented and realistic opportunity to achieve this goal.
- Diffuse parenchymal lung disease
- Genetic screening/counselling
- Genetics
- Clinical genetics
Statistics from Altmetric.com
Background
Idiopathic interstitial pneumonias (IIPs) represent a large and heterogeneous group of disorders characterised by varying patterns of inflammation and fibrosis, but often sharing similar clinical, radiological and physiological features.1 Idiopathic pulmonary fibrosis (IPF), the most common and severe of the IIPs, is a chronic, progressive and inevitably fatal lung disease of unknown origin characterised by irreversible scarring of the lung parenchyma resulting from excessive collagen production by proliferating fibroblasts/myofibroblasts.2 ,3 IPF is defined by the usual interstitial pneumonia (UIP) pattern of fibrosis, which is characterised on chest CT by subpleural, basal-predominant reticular abnormalities, traction bronchiectasis and honeycombing (figure 1), and histologically by patchy involvement of the lung parenchyma by fibrosis and micro-honeycombing in a predominantly subpleural/paraseptal distribution, and presence of fibroblastic foci (figure 2).3 The diagnosis of IPF, however, requires also the exclusion of all known causes of pulmonary fibrosis, as a UIP pattern can be found in a number of clinical contexts and aetiological conditions other than IPF, including connective tissue diseases (CTDs), chronic hypersensitivity pneumonitis and asbestosis.4

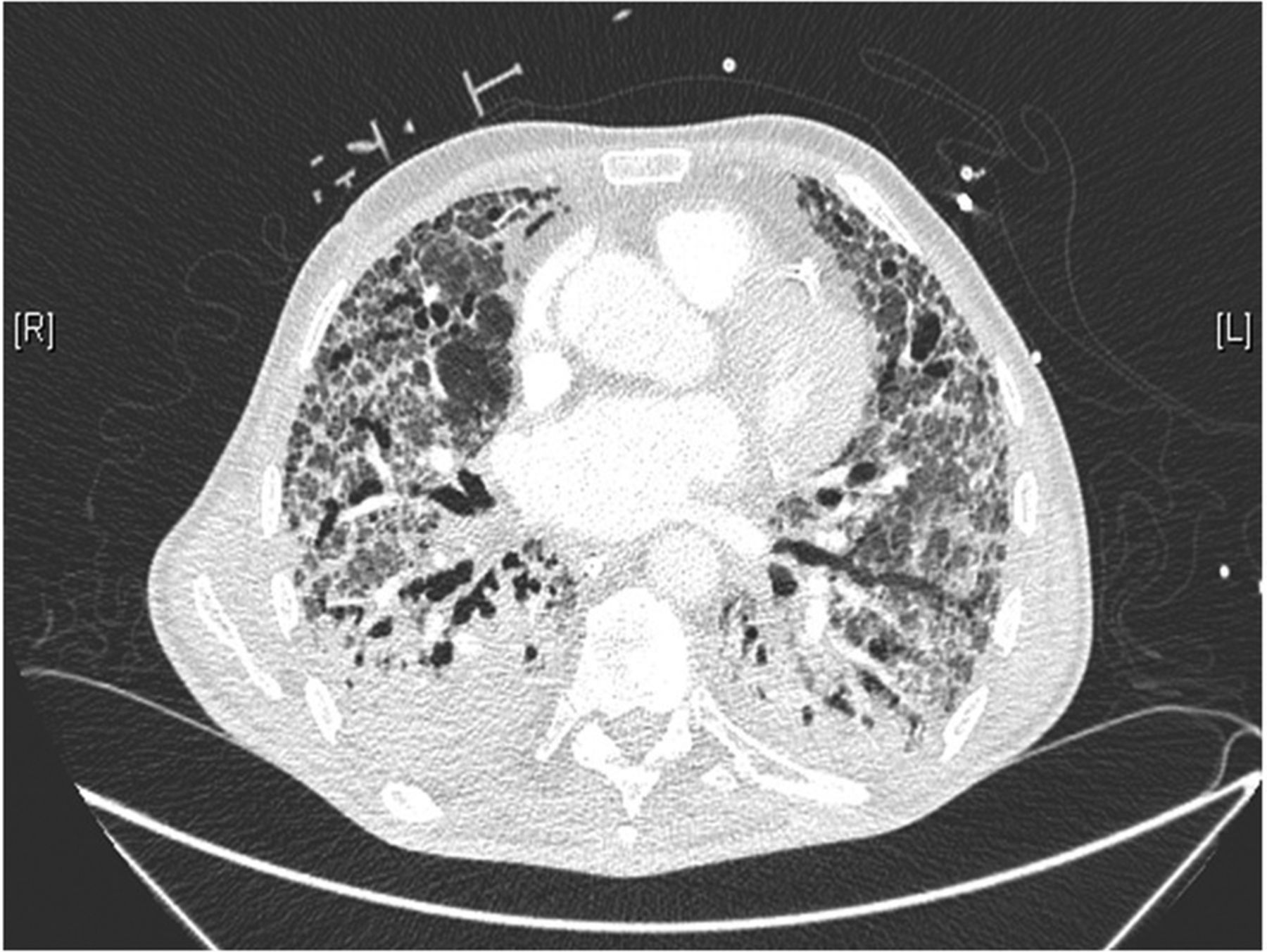
Radiological usual interstitial pneumonia. CT scan showing advanced pulmonary fibrosis characterised by cystic changes (solid arrow) and traction bronchiectasis (dotted arrow) with peripheral and lower lobe predominance.

Histologic usual interstitial pneumonia pattern. Section displaying patchy interstitial fibrosis with abrupt transition from dense fibrosis (*) and honeycomb changes (#) to relatively preserved lung parenchyma (§). H&E stain, 100× original magnification. Slide courtesy of Dr Giulio Rossi, Aosta, Italy.
With a conservative incidence range of 3–9 cases per 100 000 per year in Europe and North America, IPF is an uncommon disorder, although its prevalence rises dramatically with age.5 ,6 Cigarette smoking is a well-established risk factor for IPF, being present in about two-thirds of patients.7 While the precise origin of IPF remains elusive, the disease is believed to result from recurrent alveolar epithelial injury coupled with dysfunctional alveolar wound-healing mechanisms.8 The overall prognosis of IPF is poor (median life expectancy from time of diagnosis is approximately 3 years), but the rate of progression in individual patients is highly variable and unpredictable.9–11 There are no therapies known to prolong life of patients with IPF, apart from lung transplantation, although two compounds have proven effective in slowing down the inexorable progression of the disease12 ,13 and might eventually impact mortality.
Several lines of evidence indicate that the development of pulmonary fibrosis is, at least partially, genetically determined. They include the considerable variability in developing the disease among individuals exposed to similar concentrations of fibrogenic dusts or organic antigens; the occurrence of pulmonary fibrosis in the context of rare genetic disorders such as dyskeratosis congenita (DC),14 Hermansky-Pudlak syndrome,15 neurofibromatosis,16 tuberous sclerosis,17 Neimann-Pick disease,18 Gaucher disease19 and familial hypocalciuric hypercalcaemia20 and, more importantly, the occurrence of pulmonary fibrosis in closely related family members, including identical twins raised in different environments,21 consecutive generations in the same families22 ,23 and family members separated at an early age.24 Yet, IPF is believed to result from a complex interaction between multiple genetic and non-genetic risk factors, including cigarette smoking, infection and microaspiration of gastric content. In addition, the relative contribution of genetic and non-genetic risk factors is likely to vary among individuals.25 Linkage studies have identified several mutations associated with familial forms of IIPs (fIIPs).26 These associations point towards host defence from inhaled insults, surfactant protein processing and trafficking and telomere integrity as key mechanisms involved in the pathogenesis of the disease.27 Notably, mutations in fIIP-associated genes may also be found in a minority of patients with sporadic IPF, suggesting the existence of shared pathogenetic mechanisms.8
In this article, we summarise the evidence for pulmonary fibrosis genetic risk factors, and discuss potential clinical implications of recent genetic discoveries. Finally, we briefly discuss how rapidly evolving bioinformatics and genomic technologies may help elucidate the fundamental mechanisms of pulmonary fibrosis.
Familial pulmonary fibrosis
While most cases of IPF are sporadic, the occurrence of pulmonary fibrosis in multiple members of the same family, commonly referred to as familial interstitial pneumonia (FIP), accounts for 2%–20% of the overall cases of IIPs.26 A number of reports have suggested that FIP is inherited as an autosomal dominant trait with reduced penetrance.25 Familial and sporadic cases are clinically and histologically indistinguishable, although familial cases tend to present at a younger age and may exhibit some differences in radiological pattern.28–31 Cigarette smoking is a well-known risk factor for the development of FIP, supporting the notion that lung irritants may accentuate genetic risk and that, similar to sporadic IPF, gene-environment interactions are pivotal in initiating the disease.8 A remarkable finding is that the histopathological pattern of familial disease may differ among members of the same family. This observation suggests that different histologic types of IIPs may be related aetiologically or even pathogenically, and that additional genetic or environmental factors acting on a common genetic background may be responsible for such phenotypic heterogeneity.31 Along the same lines, subjects within families with FIP who carry telomerase gene mutations may occasionally develop chronic fibrotic hypersensitivity pneumonitis or pulmonary fibrosis associated with CTD, further suggesting gene-environment interaction.
Surfactant protein-related genes
Surfactant protein C (SFTPC) encodes surfactant protein C (SP-C), a highly hydrophobic protein that is essential for lung function and homeostasis. In 2001, Nogee et al identified a loss-of-function mutation (c.460+1 G/A) within SFTPC in a full-term baby girl with non-specific interstitial pneumonia (NSIP) and her mother who had been diagnosed with desquamative interstitial pneumonia at 1 year of age.32 In both the patient and her mother, the SFTPC mutation was identified on only one allele, consistent with an autosomal dominant pattern of inheritance. Subsequently, Thomas et al identified a heterozygous missense mutation in exon 5 of SFTPC (+128 T>A) in a large familial pulmonary fibrosis kindred, including adults with UIP and children with cellular NSIP.33 This mutation (L188Q) results in an abnormal protein precursor that accumulates in the endoplasmic reticulum (ER) and causes ER stress.33 In turn, ER stress may induce the activation of the unfolded protein response (UPR), a cascade of events that, although designed to protect the cell, may lead to alveolar epithelial cell (AEC) apoptosis in case of long-standing or severe activation.34 ,35 Severe ER stress and apoptosis of type II AEC are also present in sporadic IPF.36 A similar pathogenetic mechanism is likely to underlie the associations between mutations within surfactant protein A2 (SFTPA2) or ABCA3 and FIP.37 ,38 Subjects carrying SFTPA2 mutations have also an increased risk of developing lung cancer, which is however more linked to tobacco smoking than to family history.37
Telomerase complex genes
Pulmonary fibrosis occurs in approximately 20% of patients with DC,39 a rare inherited disorder that typically affects young males and commonly manifests as bone marrow failure, oral leukoplakia and nail dystrophy. Mutations in genes related to telomere biology and integrity have been implicated in the pathogenesis of DC.40 In 2007, two groups identified heterozygous loss-of-function mutations in TERT and TERC, which encode the two key components of the telomerase complex, in 7%–15% of FIP cases who did not have a history of DC (figures 3⇓–5).14 ,41 Telomerase mutations lead to short telomeres in both peripheral blood and in the lung,40–43 and short leucocyte telomere length is associated with worse survival in patients with IPF.44 Notably, mutations within TERT or TERC can be found also in approximately 1%–3% of sporadic IPF cases.43 While the most common histopathological pattern observed in patients carrying telomerase mutations is UIP, heterogeneous patterns of IIPs have been reported in individuals with any given mutation and in family members carrying the same mutation, indicating that short telomeres can cause a spectrum of histopathological appearances (figure 6).40 In addition to mutations in TERT and TERC, rare variants in DKC1, which encodes the telomerase complex component dyskerin, have recently been described in patients with FIP.45 ,46 Pulmonary fibrosis has been also identified in families with DC associated with mutations in TINF2.47 ,48

Patient A. Pulmonary fibrosis in a patient aged 75 years with familial premature hair greying and TERC mutation (January 2014). A sister and a twin brother are also affected by pulmonary fibrosis. Mutation analysis courtesy of Dr Caroline Kannengiesser, Paris, France.

Acute exacerbation of pulmonary fibrosis in patient A (January 2015), who died soon thereafter.

Patient B, a twin brother aged 76 years of patient A, also carrying TERC mutation (February 2015). CT scan showing reticular changes in the left lower lobe (arrow). Mutation analysis courtesy of Dr Caroline Kannengiesser, Paris, France.

Putative role for dysfunctional telomeres in the pathobiology of idiopathic pulmonary fibrosis.
More recently, rare loss-of-function mutations within RTEL1, which encodes a key regulator of telomere elongation, and PARN, which encodes an exoribonuclease involved in mRNA processing, have been found to segregate with FIP using whole-exome sequencing.49 ,50 Similar to mutations in TERT, TERC and DKC1, rare variants in RTEL1 and PARN are associated with short telomeres. However, patients with IPF and FIP have significantly shorter telomeres compared with age-matched controls in both peripheral blood leucocytes and AECs irrespective of carriage of loss-of-function mutations in telomerase genes,43 ,46 suggesting the existence of additional factors (eg, cigarette smoking and age) contributing to telomere shortening.51 In addition, it has been shown that offspring of individuals with TERT or TERC mutations have short telomeres relative to age-matched controls even if they do not carry a mutant gene,52 ,53 although it remains unclear whether the telomere shortening in these individuals is a risk factor for telomere-mediated phenotypes, including pulmonary fibrosis (box 1). Cumulatively, rare variants in telomere-related genes are present in about 15%–20% of families with FIP.26
Pulmonary and extrapulmonary features of telomere syndromes
Pulmonary manifestations
Idiopathic pulmonary fibrosis
Non-specific interstitial pneumonia
Hypersensitivity pneumonitis
Bronchiolitis obliterans organising pneumonia
Unclassifiable pulmonary fibrosis
Premature-onset emphysema
Combined pulmonary fibrosis and emphysema
Haematological manifestations
Macrocytosis
Cytopenias (mostly thrombocytopenia)
Aplastic anaemia
B, T and natural killer cell immunodeficiency
Malignancies (myelodysplastic syndromes, acute myeloid leukaemia)
Gastrointestinal manifestations
Cryptogenic liver fibrosis/cirrhosis
Nodular atrophic liver
Liver transaminase elevation
Splenomegaly
Cutaneous manifestations
Premature hair greying/loss
Nail ridging
Bone manifestations
Osteoporosis
Avascular necrosis
Increased cancer risk (mainly epithelial cancers)
Chemotherapy/radiotherapy intolerance
MUC5B
MUC5B encodes a member of the mucin family of proteins, and a major contributor to the lubricating and viscoelastic properties of whole saliva, and lung and cervical mucus.54 Using a linkage approach followed by fine mapping and case-control association, Seibold et al identified a risk locus within a cluster of gel-forming mucin genes on chromosome 11p15.5, and found that the mutant allele (T) of a single nucleotide polymorphism located in the promoter region of MUC5B (rs35705950) was carried by 38% of patients with sporadic IIPs (mostly IPF), 34% of patients with FIP and 9% of healthy controls.55 Homozygous for the MUC5B rs35705950T allele had a 20-fold increased risk of developing both sporadic and familial pulmonary fibrosis, whereas among heterozygotes the OR for sporadic and familial disease was 9 and 7, respectively. This is a rare example in the genetic epidemiology literature of a common variant with a very large genetic effect.56 The MUC5B rs35705950T allele is the strongest risk factor identified thus far, accounting for 30%–35% of the risk of developing IPF.55 ,57–60 The frequency of the mutant allele is lower among Asian patients (approximately 7% vs <2% in healthy controls), but the risk associated with its carriage appears comparable to that observed among European ancestry.61 ,62 Notably, the MUC5B association is specific for IPF,58 ,59 ,63 suggesting both a role for MUC5B rs35705950T in disease pathogenesis and the existence of major differences in genetic susceptibility across the spectrum of fibrotic lung diseases.25
The mechanisms through which increased production of MUC5B contribute to the development of pulmonary fibrosis are poorly understood. Carriage of the rs35705950T allele—a gain-of-function variant—increases MUC5B production by >30-fold even in unaffected individuals, while production of MUC5B is increased in patients with IPF irrespective of their rs35705950 genotype.55 The observations that enhanced expression of MUC5B is localised to the peripheral airspace (distal airways and respiratory bronchioles) and honeycomb cysts64 (one of the pathological hallmarks of UIP/IPF), and that the rs35705950T allele enhances expression of MUC5B in the bronchioloalveolar epithelia65 strongly support a role for this variant in disease pathogenesis. Nevertheless, due to the linkage disequilibrium (LD) patterns at the risk locus, the possibility exists that the causative variant (in LD with rs35705950) lies in the inaccessible repetitive mucin regions at 11p15.5. Despite the robustness of the association with sporadic and familial IPF, only a small proportion of carriers of MUC5B rs35705950 mutant allele in the general population will eventually develop the disease, suggesting that genetic predisposition is necessary yet not sufficient to trigger the fibrogenetic pathways that result in IPF.
Insights from genome-wide association studies
Two large genome-wide association studies (GWAS) have been conducted to date in patients with sporadic and familiar IPF. Noth et al performed a GWAS followed by two independent case-control studies in patients of European-American ancestry.57 They found that novel variants within toll interacting protein (TOLLIP; 11p15.59) and signal peptide peptidase like 2C (SPPL2C; 17q21.31) were associated with IPF susceptibility. Interestingly, individuals who developed the disease despite carrying the protective TOLLIP mutant allele of rs5743890 had an increased risk of mortality. The TOLLIP association with IPF was independent of MUC5B rs35705950, which resides at the same locus. In fact, the LD between MUC5B and TOLLIP is low and the two genes are separated by a recombinant hotspot.55 TOLLIP is an attractive candidate in IPF pathogenesis being involved in innate immune system regulation.66 However, the individual contributions or the inter-relation between these two genes in conferring disease susceptibility and modifying prognosis remain to be elucidated. Fingerlin et al performed a GWAS of 2492 patients with fibrotic IIPs (the majority of whom had IPF) and over 6000 controls.67 This study confirmed known associations with TERC, TERT and MUC5B and identified seven novel risk loci within genes involved in host defence, cell-cell adhesion and DNA repair. Recently, the same authors extended their previous work to imputed genome-wide genotypes in 1616 non-Hispanic White cases and 4683 controls followed by validation and replication in 878 cases and 2017 controls, and identified a genome-wide significant association between the human leucocyte antigen (HLA) region and fIIP.68 Notably, two HLA alleles associated with fIIP (eg, DRB1*1501 and DQB1*0602) induced differential expression of HLA genes in lung tissue, suggesting that specific immune response against unknown pulmonary targets may contribute to disease pathogenesis at least in a subset of patients. In aggregate, the genome-wide genotyped and imputed variants are estimated to account for 35% of the variability in risk of fIIP. Table 1 summarises the main genetic associations with sporadic and familial IPF.
Overview of main genetic associations with sporadic and familial idiopathic pulmonary fibrosis
From genetic association to mechanistic hypothesis
There is little doubt that genetic factors contribute substantially to the development of pulmonary fibrosis. Yet, the full implications of the reported genetic associations remain unknown. Disease-associated mutations in surfactant protein-related genes, which are exclusively expressed by type II AECs, point towards a AEC damage and loss of reparative capacity resulting from accumulation of misfolded proteins in the ER, ER stress and abnormal activation of the UPR. Notably, collagen accumulation and pulmonary fibrosis may also result from surfactant dysfunction and reduced alveolar surface tension forces, as shown in SP-C-deficient mice following intratracheal administration of bleomycin.86 Abnormalities within AEC-specific genes may explain, at least in part, why the fibrotic process in IPF is limited to the lung.
The mechanisms through which rare variants in telomerase-associated genes contribute to the pathogenesis of IPF remain also unclear. Loss-of-function mutations alter AEC turnover and repair after injury.87 In addition, TERT-deficient mice exhibit disrupted alveolar integrity88 and increased susceptibility to cigarette smoke-induced emphysema.89 However, mouse models of telomere dysfunction recapitulate only partially human disease and are, therefore, of modest utility for mechanistic studies. Similarly, the mechanisms by which gain-of-function MUC5B rs35705950T allele confers susceptibility to pulmonary fibrosis remain elusive. MUC5B variant, by virtue of greater expression or physicochemical properties of the secreted mucus, may lead to impaired clearance of inhaled toxins and microorganisms, and abnormal bacterial colonisation, as shown in MUC5B knockout mice.90 ,91 Excessive MUC5B protein may also hamper alveolar repair by either interfering with AEC-extracellular matrix interactions (resulting in failure to re-epithelialise the alveolus) or altering surfactant properties and promoting alveolar collapse.64 ,92 While increased MUC5B expression is associated with IPF, carriers of the rs35705950T allele exhibit slower rate of disease progression than non-carriers,55 suggesting that MUC5B may actually be beneficial at least in a subset of patients. In this scenario, MUC5B could either engulf and inactivate the yet-unknown fibrogenic stimuli or act as ‘second-hand surfactant’ in patients with IPF lacking surfactant, and thereby reduce alveolar obliteration and collapse.93
Taken together, these observations indicate that genetic factors, either alone or in combination with environmental exposures, contribute to make the alveolar epithelium particularly vulnerable to a multitude of insults, such as cigarette smoking, infection or chronic microaspiration of gastric content. Injured type II AECs, in an attempt to restore functional integrity, release a variety of cytokines and growth factors that promote recruitment and activation of apoptosis-resistant fibroblasts, the key mediators of fibrotic tissue remodelling. In turn, fibroblasts differentiate to myofibroblasts (a more aggressive profibrotic phenotype) and form fibroblastic foci, the histopathological distinguishing feature of UIP. This series of events leads to an exaggerated production of extracellular matrix components, tissue remodelling and architectural distortion rather than normal repair (figure 7).8 ,94

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hypothetical model of the pathobiology of idiopathic pulmonary fibrosis. AEC, alveolar epithelial cell.
Potential implications of genetic discoveries
Detection of early/subclinical disease
It is reasonable to speculate that it takes several years for pulmonary fibrosis to develop and become clinically evident, meaning that the disease can be diagnosed at a subclinical/early stage and before the lung has been extensively and irreversibly damaged.25 The term subclinical interstitial lung abnormalities (SILA) refers to the presence on chest CT scans of subtle abnormalities such as subpleural reticular changes, honeycombing, traction bronchiectasis, ground glass and centrilobular nodules.95 Similar to smokers undergoing systematic CT screening for lung cancer, SILA are also relatively common findings among asymptomatic first-degree relatives of individuals with fIIP, and accumulating evidence indicates that they may precede the development of clinically overt pulmonary fibrosis.96 ILA were identified in 177 (7%) of 2633 individuals from a general population sample who underwent chest CT as part of the Framingham Heart Study (FHS). Remarkably, for each copy of the MUC5B rs35705950T allele the odds of having ILA increased by 2.8-fold, and the risk was even greater among individuals with definite CT evidence of pulmonary fibrosis.97 A follow-up study of the same population has recently shown that 118/1867 (6%) FHS participants who had serial chest CT either developed or had progression of ILA over a 6-year period.98 Subjects with ILA progression were older, had increasing copies of the MUC5B rs35705950 mutant allele, experienced an accelerated functional decline and had an increased rate of mortality during follow-up. Notably, the prevalence of ILA is substantially higher (50–200 times) than that of IPF,97 ,99–101 suggesting that IPF represents an advanced and clinical relevant stage of a relatively common and often minimally symptomatic pulmonary syndrome that may progress at variable rates.95
Prediction of disease behaviour
The clinical course of IPF is highly variable and unpredictable:3 most patients experience a slow, yet progressive, decline in lung function that occurs over a period of years, while in 10%–15% of individuals the disease progresses from first symptoms to respiratory failure and death over a period of months.9 ,102 A third pattern of disease behaviour is characterised by periods of relatively slow decline punctuated by episodes of acute worsening (referred to as acute exacerbations) with a poor prognosis.103 While it is possible to recognise these dramatically different patterns of disease behaviour retrospectively, it is not possible, with currently available clinical tools, to predict how any given individual's disease will progress in the future.
Carriage of the mutant MUC5B rs35705950 allele is associated with the risk of developing IPF and also with a better prognosis,104 an observation that is difficult to explain. One possibility is that IPF is an aetiologically heterogeneous and biologically dynamic condition, and that different combinations of genetic and environmental factors give rise to different pathogenetic mechanisms and IPF phenotypes.104 Alternatively, MUC5B rs35705950T may increase the risk of a less severe form of pulmonary fibrosis. On the other hand, carriers of the mutant MUC5B allele are also more likely to display (or to progress to) the typical radiological manifestations of UIP/IPF (eg, reticular abnormalities with peripheral and bibasilar predominance, and subpleural honeycombing).105 A similar remarkable finding (eg, the same variant conferring opposite susceptibility and survival effects) has been observed with TOLLIP, where individuals who developed IPF despite having the protective minor allele of rs5743890 carry an increased risk of mortality.57
O'Dwyer et al evaluated the effects of a functional polymorphism (Leu412Phe) within toll-like receptor 3 (TLR3), which encodes a receptor that mediates the innate immune response to tissue injury, inflammation and viral infection,106 in primary lung fibroblasts from patients with IPF.73 They demonstrated that fibroblasts homozygous or heterozygous for the polymorphic allele exhibit a defective interferon (IFN)-β response and an enhanced fibroproliferative response after TLR3 challenge, an effect that was ameliorated in the presence of recombinant IFN-β. In patients with IPF, carriage of the mutant allele of TLR3 L412F was also associated with a significantly greater risk of mortality and an accelerated functional decline. This study indicates that the TLR3 L412F polymorphism is a potential marker of rapidly progressive disease and that defective TLR3 function may represent a potential therapeutic target, at least in a subset of patients with IPF.
Prediction of response to treatment
Oldham et al have recently published the first pharmacogenetic study in IPF using paired clinical and genotype data from patients enrolled in the ‘Effectiveness of Prednisone, Azathioprine, and N-Acetylcysteine in patients with IPF’ (PANTHER-IPF) clinical trial.107 PANTHER-IPF was a negative study. Indeed, combination of prednisone, azathioprine and N-acetylcysteine (NAC), as compared with placebo, was actually associated with a significant increase in all-cause mortality and all-cause hospitalisations,108 while NAC monotherapy was not superior to placebo in reducing the rates of decline in forced vital capacity (FVC).109 Oldham et al identified a significant interaction between NAC monotherapy and a coding variant (rs3750920) within exon 3 of TOLLIP, and showed that patients randomised to NAC who were homozygous for the CC genotype had an increased risk of disease progression (defined as a composite end point of death, transplantation, hospitalisation and ≥10% decline in FVC), whereas those homozygous for the TT genotype had a decreased risk. While the study had important limitations, mainly related to its post hoc exploratory design and the small sample size, it provides provocative data and highlights the importance of prospective, randomised, genotype-stratified clinical trials in IPF.110
Lung transplantation is the only intervention that prolongs survival of patients with IPF.3 In an international series of patients with IPF who underwent lung transplantation, carriers of loss-of-function telomerase mutations were shown to be at high risk of severe post-transplant complications, particularly bone marrow failure, reflecting the syndromic nature of their disease.111 These observations, coupled with those of a recent study from the French GERM‘O’P,112 indicate that, similar to bone marrow recipients, genetic screening for telomere syndromes may inform haematological risk and guide post-transplant management in patients with IPF.113
Genetic testing
At present, there is no recommendation on routine genetic testing for rare or common genetic variants in patients with sporadic IPF.26 ,28 In individuals with familial IPF, conversely, it is advisable to consider genetic testing for variants in surfactant protein-related genes in the presence of a family history of neonatal respiratory distress or childhood IIP, and telomerase-related genes, particularly in the presence of a family history suggestive of a telomerase dysfunction syndrome (eg, aplastic anaemia, cryptogenic cirrhosis or premature greying) (box 2). However, because of the incomplete penetrance and variable phenotypic expression of FIP-associated mutations, appropriate counselling should precede genetic testing. In addition, and more importantly, close follow-up of patients who undergo genetic testing remains essential regardless of whether they carry a disease-associated mutation.26
Settings in which genetic testing should be considered (according to authors)
Individuals with interstitial lung disease, either idiopathic or non-idiopathic, and at least one of the following:
Familial interstitial pneumonia
Idiopathic interstitial pneumonia occurring before the age of 50 years
Personal or family history of
bone marrow failure, thrombocytopenia or myelodysplastic syndromes
dyskeratosis congenita
cryptogenic cirrhosis
Modified from reference 114.
The missing heritability
Genetic factors significantly influence the development and behaviour of sporadic and familial cases of IPF. Most cases lack a clear genetic link and remain idiopathic. However, epidemiological studies suggest that genetic cases are likely to be underestimated. Possible explanations for this missing heritability include much larger numbers of common variants of smaller effect yet to be identified; rarer variants with larger effects or structural variants (insertion, deletion, duplication, translocation or inversion of segments of DNA) poorly captured by current genotyping assays; undetected gene-gene interactions or inadequate accounting for environmental factors.115 The recently developed next-generation sequencing technologies (eg, whole-genome sequencing, whole-exome sequencing, targeted region sequencing) will likely increase the number of genetic variants associated with IPF.50 ,116 Yet, in order to prove causation (beyond association), integration of genetic, gene expression as well as functional studies will be required. An alternative explanation for the missing heritability involves epigenetic mechanisms, which also have been involved in the pathogenesis of IPF.117
Epigenetic regulation of gene expression in IPF
The term epigenetics refers to any process that alters gene activity without changing the DNA sequence.118 Traditionally, epigenetic processes refer to DNA methylation, and histone modifications, although non-coding RNAs are often considered part of the epigenome. Several lines of evidence support a central role for epigenetic mechanisms in IPF.119 First, IPF is a disease of the elderly and profibrotic changes in DNA methylation occurring with age may predispose to the development of the disease in susceptible individuals.120 Second, IPF is more common among smokers and cigarette smoking influences DNA methylation in genes involved in the pathogenesis of the disease.121 Third, epigenetic mechanisms are essential in lung development, and aberrant recapitulation of developmental programmes is an hallmark of IPF.122 Finally, molecular processes highly relevant to pulmonary fibrosis, such as fibroblast apoptosis123 and cell senescence124 have been shown to be epigenetically regulated. Genome-wide evaluation of epigenetic tags in IPF lung tissue have identified extensive DNA methylation changes.125 Yang et al have recently performed the most comprehensive study of IPF lung tissue to date;126 this study confirmed that the DNA methylation status is altered in IPF and suggested a substantial effect of altered DNA methylation profile on gene expression.
Conclusions
In IPF, the genetic associations identified thus far have broadened our understanding of disease pathogenesis, suggesting a prominent role for AEC injury, host defence and telomere integrity. Identification of genetic signatures that predict disease behaviour and response to treatment (eg, theragnosis) would enable tailoring of therapy (or referral for lung transplantation) and appropriate prescribing of drugs, thus reducing costs and unnecessary adverse events. In clinical trials of IPF, a major obstacle is the heterogeneity of the patient population, where the rate of disease progression is not uniform. A genotype-guided enrolment has the potential to identify compounds that are effective in selected groups of patients. A personalised approach to treatment is successfully used in oncology. There is genuine hope that a similar approach may prove equally successful in IPF.
References
Footnotes
Contributors PS conceived of the study and drafted the manuscript. VC drafted the manuscript and revised it for important intellectual content. Both authors read and approved the final manuscript.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.