Article Text

Abstract
Pulmonary arterial dissection is an extremely rare and usually lethal complication of chronic pulmonary hypertension. The condition usually manifests as cardiogenic shock or sudden death and is therefore typically diagnosed at postmortem examination rather than during life. However, recent isolated reports have described pulmonary artery dissection in surviving patients. The first case of pulmonary artery dissection in a surviving patient with cor pulmonale caused by chronic obstructive pulmonary disease is presented. The aetiology, pathophysiology, and clinical presentation of pulmonary artery dissection are reviewed and factors that may aid diagnosis during life are discussed.
- pulmonary artery dissection
- cor pulmonale
Statistics from Altmetric.com
Although dissection of the systemic arteries is a well recognised and often non-fatal consequence of essential hypertension, pulmonary arterial dissection is an extremely rare and usually lethal complication of chronic pulmonary hypertension. Pulmonary artery dissection has most commonly been described in patients with underlying congenital heart disease and in those with primary pulmonary hypertension. The condition usually manifests as cardiogenic shock or sudden death and is therefore typically diagnosed at postmortem examination rather than during life.1–4 However, recent isolated reports have described pulmonary artery dissection in surviving patients. Despite the relatively common causal association of chronic obstructive pulmonary disease with pulmonary hypertension, pulmonary artery dissection has rarely been reported in this group of patients. We present the first case of pulmonary artery dissection in a surviving patient with cor pulmonale caused by chronic obstructive pulmonary disease. We also review the aetiology, pathophysiology, and clinical presentation of pulmonary artery dissection and discuss factors that may aid diagnosis during life.
CASE REPORT
A 56 year old woman presented to the emergency department with notable shortness of breath and mild bilateral leg oedema. About 30 years previously, she had had extensive pulmonary tuberculosis treated with streptomycin. Since then she had experienced mild dyspnoea on exertion but over the preceding six months her exercise capacity had reduced greatly to about 50 m. Three weeks before admission, she had developed a productive cough and malaise. She had also noticed an isolated episode of a severe, sharp pain in the upper back radiating to the left anterior chest and arms associated with sweating and lasting for about 10 minutes. At that time she was empirically treated with antibiotics resulting in resolution of the cough, but the dyspnoea continued to worsen and was accompanied by the development of peripheral oedema. She was a longstanding heavy smoker; she did not have any pets, and there was no history of occupational dust exposure. Clinical examination showed a thin body habitus, respiratory rate of 22 breaths/min, resting tremor, peripheral cyanosis, palmar erythema, and mild kyphoscoliosis. She was afebrile, her heart rate was 100 beats/min, and her blood pressure was 145/92 mm Hg. The jugular venous pressure was raised and there was bilateral leg oedema to the knees. A right ventricular heave was palpable and auscultation found a loud pulmonary component of the second heart sound followed by an early diastolic murmur heard loudest over the left sternal edge radiating to the pulmonary area and apex. Chest expansion and breath sounds were greatly reduced bilaterally. A smooth liver edge was palpable below the right costal margin but there was no evidence of ascites.
Full blood count, serum urea and electrolyte concentrations, liver function tests, thyroid function tests, and C reactive protein concentration were all normal. Arterial blood gases on air had a pH of 7.41, Po2 6.5 kPa, Pco2 8.0 kPa, HCO3− 40.3 mmol/l, and O2 saturation of 87%. Chest radiography was reported as showing enlarged pulmonary arteries with loss of lung volume and calcification at the apices consistent with previous tuberculosis. Her 12 lead ECG showed sinus rhythm with ventricular ectopic activity, P pulmonale, right axis deviation, and dominant R waves in V1. Pulmonary function tests showed a severe obstructive respiratory pattern with forced expiratory volume in one second (FEV1) of 0.48 l, forced vital capacity of 1.18 l, and a ratio of 41%. Echocardiography showed a dilated, severely hypokinetic right ventricle with flattening of the interventricular septum suggestive of raised right ventricular pressure. The right atrium was also dilated with mild tricuspid regurgitation and an estimated pulmonary artery systolic pressure of about 75–80 mm Hg. The main pulmonary trunk was greatly dilated at 6 cm and there was mild to moderate pulmonary regurgitation. A dissection flap with bidirectional flow was noted in the main pulmonary trunk originating just above the pulmonary valve and extending to the bifurcation of right and left pulmonary arteries, adjacent to the origin of the left pulmonary artery (figs 1 and 2). Subsequent computed tomography (CT) of the thorax with pulmonary angiography confirmed generalised ectasia of the main pulmonary trunk with the appearance of a linear line consistent with dissection on axial sections and sagittal reconstruction (fig 3). There was no evidence of haematoma formation.
The patient was treated with 28% oxygen and intravenous loop diuretics, which rapidly resolved the peripheral oedema. Her arterial blood gases after overnight 28% oxygen had a pH of 7.37, Po2 16.5 kPa, Pco2 8.6 kPa, and O2 saturation of 96%. In view of the high anaesthetic risk related to the severely reduced ventilatory reserve, surgical repair of the pulmonary artery dissection was not considered a viable option. Her continued treatment is long term domiciliary oxygen, diuretics, and angiotensin converting enzyme inhibitor and she remained stable at 12 months’ follow up.
DISCUSSION
This report describes the first case of pulmonary artery dissection with underlying severe pulmonary hypertension caused by chronic obstructive pulmonary disease in a surviving patient. Pulmonary artery dissection was manifest by the development of chest pain, worsening dyspnoea, and right heart failure. On the basis of the clinical history, pulmonary artery dissection may have been precipitated by a community acquired chest infection leading to worsening hypoxia, thereby further raising pulmonary artery pressure. The diagnosis was reliably made by transthoracic echocardiography, providing excellent delineation of the origin and distribution of the intimal flap; subsequent CT confirmed the diagnosis. There was generalised dilatation of the main pulmonary trunk and branch arteries caused by chronic pulmonary hypertension but no evidence of localised aneurysm formation.

Transverse section of a computed tomographic pulmonary angiogram showing a linear dissection (arrow) of the main pulmonary trunk.
Over the past two centuries, 63 other cases of pulmonary artery dissection have been described,1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25 of which only eight were diagnosed during life.18–25 Consequently, an appreciation of the anatomical abnormalities and pathological processes leading to pulmonary artery dissection has mainly arisen from postmortem findings. The majority of pulmonary artery dissections occur in the presence of medial degeneration with fragmentation of elastic fibres and generalised dilatation of the pulmonary arterial tree caused by chronic pulmonary hypertension.1–3,23 With medial degeneration, the wall is weakened, the vessel may dilate, and the raised intravascular pressure and shear stresses may predispose to the development of an intimal tear. Whether medial degeneration causes the dissection, predisposes to intimal tears, or results from chronically raised intravascular pressure remains controversial. The main pulmonary trunk is the site of dissection in about 80% of cases, usually without involvement of the branches; the remainder of cases are isolated to the right or left pulmonary arteries and intrapulmonary branches.1–3,23 In a small proportion of cases, pulmonary artery dissection may occur at the site of localised aneurysm formation. Aneurysms are most commonly associated with congenital heart lesions that have led to sustained high pulmonary flow rates and pulmonary hypertension, in particular persistent ductus arteriosus.21 In rare cases, aneurysms are associated with infection, inflammation, trauma, atherosclerosis, or connective tissue disorders, often in the absence of pulmonary hypertension. In contrast to aortic dissection, the false lumen in pulmonary artery dissection tends to rupture rather than to extend distally and develop a re-entry site.1–3,23 Rupture occurs most commonly into the pericardium and rarely into the lungs, mediastinum, or pleural cavity, leading to sudden death. Of 50 postmortem examinations in which the mechanism of death of the patient was stated, rupture leading to cardiac tamponade was identified in 39.23
Among the 63 previously documented cases of pulmonary artery dissection, 34 patients had underlying cardiac disease (25 congenital heart defects, most commonly persistent ductus arteriosus, and nine rheumatic mitral stenoses), nine had primary pulmonary hypertension, four had pulmonary thrombosis, four had chronic obstructive pulmonary disease, two had catheter induced dissection, three had idiopathic dissection, three had inflammatory causes, and four had an unspecified cause. There was a roughly equal sex distribution. The ages ranged from 26 days to 85 years with incidence peaks in the third and sixth decades. In younger patients, congenital heart abnormalities were the most common underlying causes, whereas in older patients a variety of acquired diseases were associated with pulmonary artery dissection. Interestingly, eight cases were documented to occur in the absence of chronic pulmonary hypertension and had idiopathic, inflammatory, or catheter induced causes.
Over the past two decades, pulmonary artery dissection has been diagnosed during life in eight previous cases.18–25 This may partly reflect the technological development and increasing use of sophisticated non-invasive cardiac imaging modalities during this period. Non-invasive imaging methods were used to detect pulmonary artery dissection in seven of the eight patients. Echocardiography, CT, and magnetic resonance imaging (MRI) detected pulmonary artery dissection in three of seven patients, three of four patients, and two of three patients, respectively, in whom these techniques were used. In one case, non-invasive imaging was not used and the diagnosis was made from pathological specimens.23 Differences in the relative diagnostic abilities of echocardiography, CT, and MRI may be related to variability in individual anatomy, localisation and distribution of the intimal flap, and inherent limitations of the imaging techniques. Nevertheless, these cases show that in any given patient, single or multiple non-invasive imaging methods may reliably be used to detect the presence of pulmonary artery dissection. Echocardiography appears to be the logical first line investigation because of its ready accessibility, ease of use, and low cost; CT or MRI may be used if echocardiography fails to detect an intimal flap or to provide complementary information related to the dissection and associated abnormalities. Among the live patients, chest pain and dyspnoea were the most common presenting symptoms. Three patients had secondary pulmonary hypertension caused by congenital or valvar heart disease,18,21,25 three had primary pulmonary hypertension,19,22,24 and two had evidence of pulmonary thrombosis20,23 as the underlying disease process. Surgical repair was successful in three of these patients.21,22,24
The reasons for the infrequent association of pulmonary artery dissection with chronic obstructive pulmonary disease compared with congenital heart defects and primary pulmonary hypertension are unclear. These may partly relate to the different pathophysiological mechanisms leading to the development of pulmonary hypertension in these separate disease entities. Chronic obstructive pulmonary disease predisposes to pulmonary hypertension mainly by promoting hypoxia induced vasoconstriction rather than causing direct pulmonary vascular damage. In contrast, congenital heart defects and primary pulmonary hypertension cause intrinsic pathological abnormalities within the pulmonary vasculature that may increase the likelihood of vessel wall injury. Congenital heart defects cause haemodynamic alterations leading to increased pulmonary blood flow, raised intravascular pressure, and increased wall stress; primary pulmonary hypertension is associated with morphological manifestations of vascular injury, coagulation disturbance, and inflammatory effects within the pulmonary circulation. It is possible, though difficult to prove, that the previous tubercular infection in our patient may have played a part in predisposing to the dissection by promoting chronic inflammatory changes to the pulmonary arterial tree leading to a weakened vessel wall.
Summary
Pulmonary artery dissection is a rare and life threatening condition that should be suspected in patients with chronic pulmonary hypertension presenting with chest pain, worsening dyspnoea, or haemodynamic compromise. Although pulmonary artery dissection is classically diagnosed at postmortem examination, the advent of high quality non-invasive imaging methods in recent years has had a central role in detecting this condition during life. Moreover, recent reports have described successful surgical repair of pulmonary artery dissection in isolated cases. These findings necessitate an increased awareness of the condition with a view to early diagnosis and corrective intervention in selected cases.
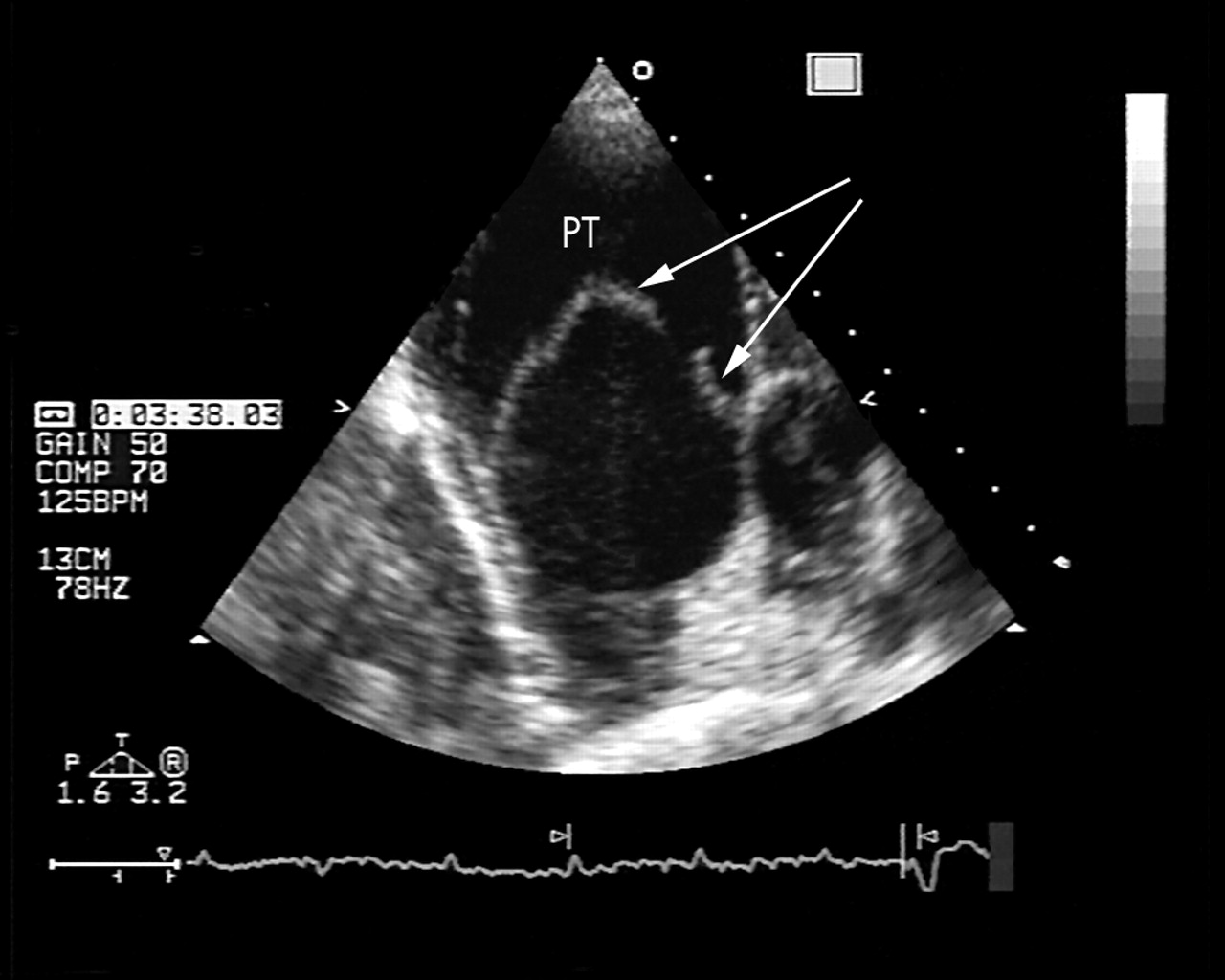
Two dimensional transthoracic echocardiogram in a modified short axis view showing a greatly dilated pulmonary trunk (PT) with an intimal flap in transverse section.

{kind=link}
{kind=link}
{kind=link}
Transthoracic echocardiogram showing longitudinal distribution of the intimal flap (arrow) originating from just above the pulmonary valve and extending to the origin of the left pulmonary artery (LPA). The presence of bidirectional flow on colour flow imaging confirms the presence of dissection with a true and false lumen. PA, pulmonary artery; RPA, right pulmonary artery.