





Abstract
The present review aims to examine the effect of specific drugs on long-term outcome of pulmonary arterial hypertension (PAH), to critically review the available data, and to derive useful information for daily patient care.
PAH is an intrinsic disease of the pulmonary circulation with a malignant evolution as a consequence of progressive right heart failure. Without specific therapy, median survival is only 2.8 yrs. The intravenous prostacyclin analogue epoprostenol is the only treatment with a demonstrated effect on survival, observed during a single 12-week randomised placebo-controlled trial. Three long-term observational studies have also shown that median survival is raised above 6 yrs with this therapy. Subcutaneous treprostinil appears to have similar beneficial effects on survival, as reported in two long-term observational studies. This is not the case for inhaled iloprost, as shown in one study in which a high proportion of patients needed the addition of, or the switch to, another therapy. Among the oral agents, long-term data have only been published for bosentan. The three studies including patients from expert centres also showed very good survival data, but again with a broad use of combination therapy. In less expert hands, with limited access to more complex therapies, reported survival seems much worse. In these studies, baseline New York Heart Association class and 6-min walk distance are repeatedly shown to be important predictors of survival. Finally, there is emerging data that prostanoid therapy results in a tendency to normalise C-reactive protein levels, a factor associated with improved long-term outcomes.
Pulmonary arterial hypertension (PAH) is an intrinsic, pre-capillary pulmonary arteriopathy characterised by intima fibrosis, media hypertrophy, matrix proliferation and plexogenic lesions. It includes an idiopathic (IPAH) and a familial/inherited form, as well as pulmonary hypertension associated with connective tissue disease, congenital systemic-to-pulmonary shunts, portal hypertension, HIV and the use of drugs or anorexigens.
In the pre-prostacyclin era, the median survival of untreated patients with IPAH was 2.8 yrs [1], placing IPAH among the most malignant diseases of the middle-aged individual. The 12-week epoprostenol pivotal trial was the first step forward, showing a lower death rate in patients treated with epoprostenol compared with placebo [2]. However, none of the more recent randomised controlled trials have been able to confirm this trend. However, by combining these trials in a meta-analysis involving a total of 3,140 patients, a significant mortality reduction was recently demonstrated [3].
Current PAH-specific therapies target three pathways: the 1) prostacyclin and 2) nitric oxide pathways, with cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) as respective second messengers, both acting as vasodilators and inhibitors of smooth muscle cell proliferation; and 3) the endothelin (ET)-1 pathway, acting via a G protein-coupled receptor as a vasoconstrictor and a proliferating agent. Prostacyclin analogues include epoprostenol, exclusively used intravenously because of its very short half-life, and more stable compounds, such as treprostinil, which can be used subcutaneously, intravenously, inhaled or orally, iloprost for inhaled and i.v. use, and beraprost for oral use. Inhibitors of cGMP phosphodiesterase 5, such as sildenafil and the longer-acting tadalafil, are orally administrated and used to increase intracellular concentrations of cGMP. The selective ETA- (sitaxsentan and ambrisentan) and dual ETA- and ETB-receptor (bosentan) antagonists have been developed for oral use.
The regulatory approval and subsequent clinical use of these agents is based on trials of between 12 and 16 weeks’ duration demonstrating improvements in exercise capacity. The present review discusses current evidence exceeding this time interval. Given the paucity of randomised trials, conclusions are more difficult. We ask, therefore, do we really improve long-term survival in PAH? And, if so, what may be the optimal strategy?
LONG-TERM PLACEBO-CONTROLLED STUDIES
The only published long-term, placebo-controlled study in PAH is the American beraprost study [4]. It was a 1-yr, multicentre, double-blind, randomised, placebo-controlled trial, which included 116 PAH patients and looked at disease progression, 6-min walk distance (6MWD), peak oxygen consumption, Borg dyspnoea score, haemodynamics, symptoms and quality of life. Baseline characteristics of the study population were a mean age of 42±2 yrs, a 6MWD of 439±11 m and a New York Heart Association (NYHA) functional class II to III repartition of 52.5 to 47.5%. Beraprost was administrated four times daily at an increasing mean dose of 71 μg at 3 months, 92 μg at 6 months and 107 μg at 12 months. In this study, the initial improvement in exercise capacity was no longer significant at 1 yr. The beraprost-treated patients had improved 6MWD by 22 m from baseline at 3 months (p = 0.010) and by 31 m at 6 months (p = 0.016) compared with placebo, but not at 9 or 12 months. There was also no effect on haemodynamics at 12 months.
Beside this, some medium-term data for bosentan-treated patients exists. A 48 patient subset of the BREATHE-1 (Bosentan Randomized Trial of Endothelin Antagonist Therapy) 16-week pivotal trial [5] entered a second blinded period of 12 weeks. In the 35 patients who continued bosentan blinded treatment, there was a slight decrease in 6MWD at 28 weeks that, even if it was not significant, was somewhat worrisome (unpublished data).
Instead of increasing placebo-controlled study duration, randomised withdrawal is another way to show durability of drug effect. This approach, somewhat controversial for a life-sustaining therapy, has been cautiously used for epoprostenol, showing that down-titration after a mean of 3.4 yrs of treatment was uniformly accompanied by clinical deterioration [6].
OBSERVATIONAL STUDIES
To compensate for the scarcity of long-term controlled data on drug efficacy in PAH, large observational studies have been proposed in order to evaluate long-term outcome. These studies are presently available for most of the specific PAH medications (epoprostenol, treprostinil, iloprost and bosentan). However, we have to be aware of the numerous limitations of these studies: they were open-label studies, and there were patient drop-outs, treatment changes and an absence of contemporary control groups.
Focusing on long-term data with i.v. epoprostenol, we identified three major studies (table 1⇓). The first, by Barst et al. [7], is an open-label follow-up of 18 patients previously included in the initial randomised controlled trial from Rubin et al. [10]. The second, by Sitbon et al. [8], and the third, by McLaughlin et al. [9], are much larger single-centre series of patients, from the French and US reference centres in Clamart and Chicago, IL, respectively. In the studies by Barst et al. [7] and Sitbon et al. [8], survival of epoprostenol-treated patients was compared to a matched historical cohort, while in the study by McLaughlin et al. [9] survival was predicted from the d'Alonzo equation, derived from the US National Institutes of Health (NIH) registry population [1]. This equation has been largely used to calculate survival on the basis of haemodynamic parameters (mean pulmonary arterial pressure, cardiac index and right atrial pressure). These three studies included IPAH patients with severe disease, as evidenced by their poor cardiac index <2 L·min−1·m−2, poor baseline 6MWD of ∼250 m and the almost exclusive NYHA class III and IV inclusion. As a consequence of the well-known tolerance to prostanoid, epoprostenol dose was progressively increased to 30 ng·kg−1·min−1 at 3 yrs in the French cohort and 50 ng·kg−1·min−1 in the US series. Survival data at 1, 2 and 3 yrs are reported in table 2⇓. Barst et al. [7] reported a significant survival improvement in epoprostenol-treated patients when compared with the historical control group (p = 0.045), and the median survival reached more than 6 yrs. These results were confirmed in the Sitbon et al. [8] and McLaughlin et al. [9] series, in which survival was similar despite the substantial differences in dosage. Both studies also confirmed the importance of the baseline NYHA functional class in predicting the outcome of epoprostenol-treated IPAH patients, as originally shown in the NIH registry untreated population [1]. However, despite the poorer prognosis, median survival in class IV patients was prolonged from 6 months to >3 yrs in both cohorts.
Long-term response to intravenous epoprostenol
Long-term survival in pulmonary arterial hypertension (PAH)
More recently, two retrospective studies using subcutaneous treprostinil in patients with PAH have been published (table 3⇓). The first, by Lang et al. [14], described 122 patients, including 99 with PAH and 23 with chronic thromboembolic pulmonary hypertension, started with treprostinil in three European centres. The second, by Barst et al. [13], assessed 860 PAH patients, including patients previously enrolled in the pivotal trials as well as de novo patients from the USA. At baseline, patients had less severe disease than those of the epoprostenol series, as demonstrated by their baseline 6MWD of ∼300 m and cardiac index >2 L·min−1·m−2, the predominance of NYHA class III patients and the inclusion of some class II patients. Within 3 yrs, the treprostinil dose was progressively increased to 40 ng·kg−1·min−1 in both the European and the predominantly US cohorts. Survival data at 1, 2 and 3 yrs are reported in table 2⇑. Lang et al. [14] reported a 65-m increase in 6MWD and an improvement in NYHA class at 1 yr, both of which were maintained at 2 and 3 yrs. This contrasts with the modest improvements seen in the pivotal 12-week study [23], in which the median placebo-corrected increase in 6MWD was only 16 m, reflecting the significant dosing limitations which resulted from local infusion pain. In addition, survival among the 32 IPAH patients was significantly better than predicted and comparable with the survival obtained with i.v. epoprostenol. The favourable long-term data were confirmed in the 332 IPAH patients of the study by Barst et al. [13], showing that survival of patients receiving treprostinil in monotherapy was not different from that of the entire cohort. Oral therapy, predominantly bosentan, but also sildenafil, was added in 18% and 15% of the Lang et al. [14] and Barst et al. [13] studies, respectively. Drop-out due to infusion site pain was observed in 5% of the patients included in the Lang et al. [14] study and in 23% of the patients included in the larger cohort studied by Barst et al. [13], highlighting the importance of dedicated centres for efficient patient support. Barst et al. [13] also confirmed the predictive value of baseline NYHA functional class, as well as baseline 6MWD, pulmonary vascular resistance and mixed venous oxygen saturation, in the outcome of treprostinil-treated PAH patients. Having a baseline NYHA class IV, a 6MWD <295 m, an indexed pulmonary vascular resistance >30 WU·m−2, or a mixed venous oxygen saturation <55%, was associated with a 52–59% risk of dying within 3 yrs [24].
Long-term response to subcutaneous treprostinil
Evidence for the use of the prostacyclin-analogue iloprost is reliant upon two long-term studies: one by Opitz et al. [15], performed with inhaled administration and one by Hoeper et al. [16] with i.v. administration. In most cases, i.v. administration became a second-line therapy for patients initially treated with inhaled iloprost for a median of 12 months (table 4⇓). Whereas baseline 6MWD was not available in the cohort studied by Opitz et al. [15], most of the patients included were in NYHA class II and III. Iloprost (100 μg) was administered as six inhalations per day. In the cohort studied by Hoeper et al. [16], patients were severely sick at the start of i.v. iloprost therapy, with a baseline 6MWD of 228 m, a cardiac index <2 L·min−1·m−2 and a majority in NYHA class IV. Iloprost dose was increased to 2.6 ng·kg−1·min−1 at 6 months. Survival data at 1, 2 and 3 yrs are reported in table 2⇑. In the study by Opitz et al. [15], only 20% of the patients were still on iloprost monotherapy at 3 yrs. In the study by Hoeper et al. [16], median survival was only 3 yrs from diagnosis and 1 year from i.v. iloprost initiation, which does not differ from survival without specific treatment [1]. However, this study included a selection of patients with a mean pulmonary vascular resistance >1,500 dyn·s·cm−5, who were doing poorly on inhaled iloprost, and who were probably underdosed with i.v. iloprost (dosage used in UK centres is about 3.3 and 4.7 ng·kg−1·min−1 at 3 and 12 months, respectively). Nevertheless, this study confirmed the importance of baseline 6MWD and mixed venous oxygen saturation in predicting the outcome of iloprost-treated patients with PAH.
Long-term response to inhaled and intravenous iloprost
Concerning oral agents, long-term data are only available for the dual receptor antagonist bosentan (table 5⇓). Three series have been published: the one studied by McLaughlin et al. [17] is an open-label follow-up of patients included in two randomised controlled trials [5, 25]; those published by Hoeper et al. [18] and Provencher et al. [19] are single-centre series from Germany and France, respectively. The McLaughlin et al. [17] and Provencher et al. [19] studies only included IPAH patients. Patients were less sick than in the prostanoid studies, with baseline 6MWD 300–350 m, and predominantly in NYHA class III. They received the recommended dose of bosentan (125 mg b.d.). Survival data at 1, 2 and 3 yrs are reported in table 2⇑. McLaughlin et al. [17] reported survival that was better than predicted estimates, with a mortality of 5.5% per annum. However, 30% of the patients were on combined therapy at 3 yrs. Similarly good survival data were confirmed in the two other cohorts. The goal-oriented approach adopted by Hoeper et al. [18], with additional drugs added every 2 to 6 months if pre-defined goals were not attained (6MWD >380 m, peak oxygen uptake >10.4 mL·min−1·kg−1 and peak systolic blood pressure >120 mmHg), did not provide a survival advantage when compared to the other two series. However, in the cohort studied by Provencher et al. [19] only 40% of the patients remained on bosentan monotherapy at 3 yrs, a prostanoid being added in patients with NYHA class >II, 6MWD <250 m and cardiac index <2.2 L·min−1·m−2. Only 3% of the patients stopped bosentan because of elevated liver enzymes in this cohort.
Long-term response to oral bosentan
COMPARISON OF THE DIFFERENT THERAPEUTIC APPROACHES
Since no direct comparison between PAH-specific medications has been performed, it is interesting to place in perspective the survival data collected from the aforementioned long-term observational studies (table 2⇑). The first two rows display series performed when no PAH-specific therapy was available in the USA [1] and in China [11]. It should be emphasised that the historical data obtained by d'Alonzo et al. [1], frequently contested as a comparator, were reaffirmed by more recent survival data from China where PAH-specific therapies are not widely available. The third row displays the most recent survival data concerning lung transplantation in PAH reported by the International Society of Heart and Lung Transplantation [12].
Concerning the survival benefit raised by the PAH specific therapies, it is notable that different series across different countries or continents provided remarkably similar survival data within each drug category: 63% at 3 yrs for epoprostenol, 71% for treprostinil, 59% for iloprost and 80% for bosentan (table 2⇑). It is also important to note that: 1) these positive survival data were actually obtained with broad use of combination therapies, especially concerning bosentan studies; 2) in comparison with the NIH historical group of patients, anticoagulants were used in a larger extent in the most recent series; and 3) less sick patients were included in the bosentan series, as demonstrated by the cardiac index, NYHA class and the 6MWD. This evolution precludes the comparison of oral agent and parenteral prostanoid efficacy, although it is obvious that more patients are surviving on monotherapy with parenteral prostanoids (85%) then with oral agents (55%).
Despite being acceptable when compared with historical controls, survival rates with PAH-specific drugs seem inferior when those patients with severe symptoms are compared with results after atrial septostomy [20]. However, these data need to be confirmed by other studies. Moreover, the small group of patients with reversible IPAH (∼7%) who are long-term responders to calcium channel blockers seem to have a favourable prognosis, with survival >87% at 3 yrs [21, 22] without the need for additional, more specific, agents.
REAL-LIFE EXPERIENCE WITH PAH THERAPIES
Beside these optimistic data, another perspective on current therapeutic practices in PAH has been provided by Accredo, a company which delivers different PAH-specific drugs to US patients. They reported, at the 2008 congress of the Pulmonary Hypertension Association, that an elevated proportion of patients who were dying from PAH were on oral agent monotherapy [26]. In a series of 821 patients, who were initiated on bosentan between October 1 and December 31, 2004, overall survival at 3 yrs was only 64%, in comparison with the previously reported 80% by the German and French expert centres [18, 19]. From the 190 patients who died, 169 (89%) were on bosentan monotherapy, and only 11% were treated with prostanoids before death. The vast majority of them had never been referred to an expert centre.
The good survival data of patients treated with bosentan in expert centres were obtained with a broad and undelayed use of combination therapy including a prostanoid [18, 19]. In less experienced hands, with limited access to combined therapies, reported survival appears severely impaired.
ANTI-INFLAMMATORY EFFECTS OF PAH DRUGS AND LONG-TERM SURVIVAL
Another appealing observation is the effect of prostanoids on systemic inflammation. Prostacyclin analogues interact in different ways with inflammatory cells [27]. However, the relevance of this for the treatment of pulmonary hypertension remained unknown until the following findings were reported: 1) circulating C-reactive protein (CRP) levels are increased in PAH patients and are significantly higher in NYHA class III–IV patients and in nonsurvivors; 2) patients with CRP levels >5.0 mg·L−1 have a significantly lower survival rate and CRP is an independent predictor of survival; and 3) patients normalising their CRP levels under treatment, assigned as responders, have a significantly higher survival rate (fig. 1a⇓) [28]. It is noteworthy that the proportion of patients treated with a prostacyclin analogue was significantly higher among the responders (55% versus 17%; fig. 1b⇓). Patients undergoing prostanoid therapy also displayed significantly lower CRP levels (fig. 1c⇓).

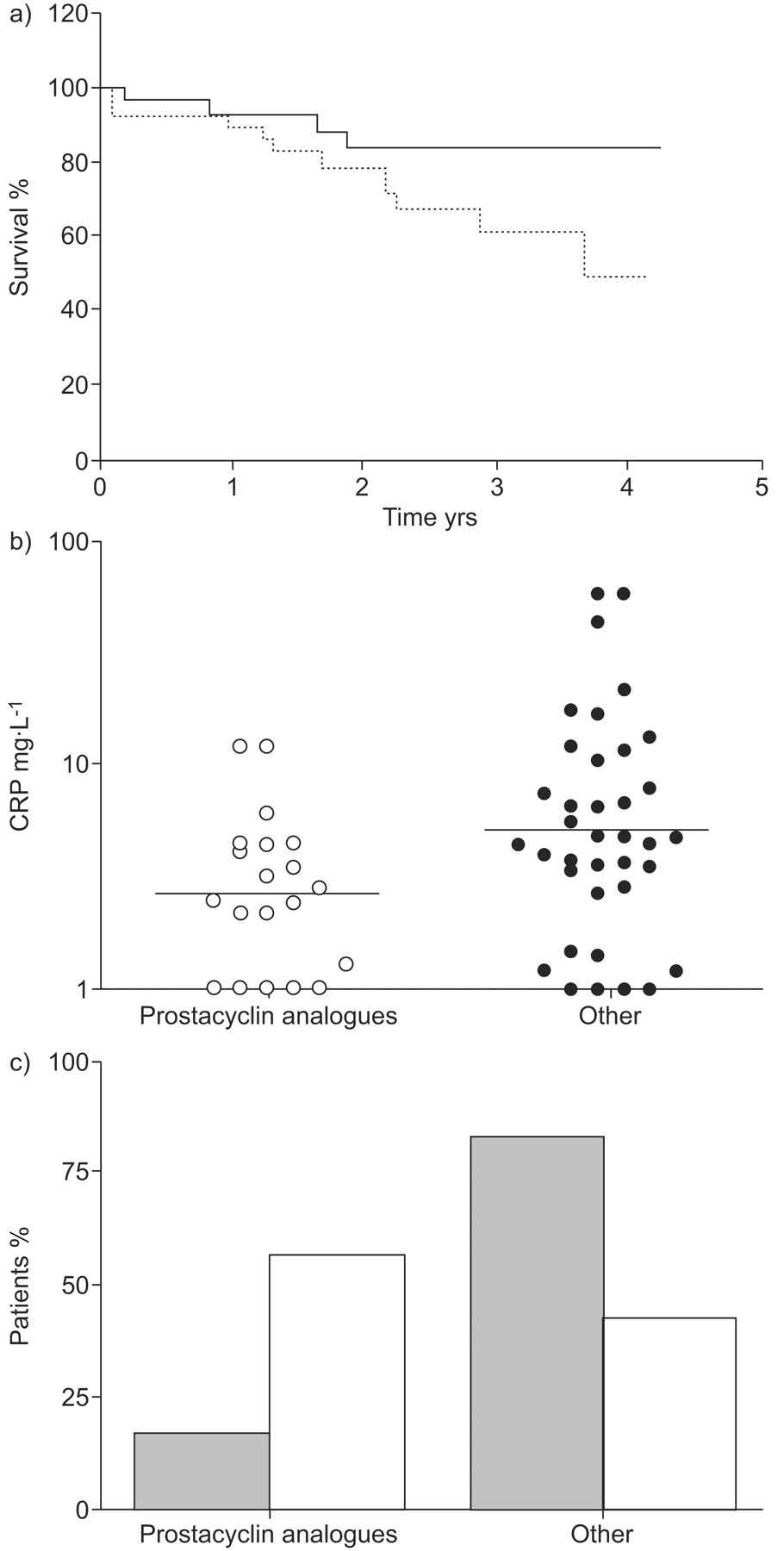{kind=link}
Effect of pulmonary arterial hypertension-specific treatment on systemic inflammation. a) Kaplan–Meyer survival curves for patients normalising their C-reactive protein (CRP) levels under treatment (responders, i.e. CRP ≤5 mg·L−1; ––––) versus nonresponders (i.e. CRP ≥5 mg·L−1; ······) (p<0.05). b) CRP levels in patients treated with prostacyclin analogues versus patients treated with another drug (p = 0.01). ––––: geometric means. c) Proportion of patients treated with prostacyclin analogues versus another drug among the responders (□) and nonresponders (▓) (p = 0.002). Reproduced from [28] with permission from the publisher.
Since inflammation appears to play a role among the pathological mechanisms of PAH [29], the anti-inflammatory properties of prostanoids, illustrated by a tendency to normalise CRP, may be a relevant finding.
CONCLUSION
In conclusion, there is a large body of very consistent evidence for improved survival from long-term observational studies performed in different centres and countries, accompanied by a sustained benefit in 6MWD and NYHA functional class. The latest series on oral therapy included patients with less severe disease and more stable prevalent cases, which artificially contributed to improved survival. Additionally, it seems that: 1) more patients are surviving on monotherapy with parenteral prostanoids than with oral agents; 2) a large proportion of patients who are dying are on monotherapy with oral agents and not treated in reference centres with access to all therapies; and 3) prostanoids seem to have a more pronounced anti-inflammatory effect, with implications for the prognosis.
This emphasises the need to escalate treatment rapidly, to promote treatment strategies including prostanoid use, and to accelerate transfer to reference centres.
Statement of interest
M. Delcroix has been on advisory boards for Actelion, GSK and Pfizer, has received speaker fees from Actelion, Encysive and LungRx, and research grants from Actelion, Encysive and GSK.
Provenance
Submitted article, peer reviewed.
Acknowledgments
The authors thank A. La Gerche (University Hospitals of Leuven, Katholieke Universiteit Leuven, Leuven, Belgium) for valuable editorial comments.
- Received May 27, 2009.
- Accepted August 10, 2009.
- © ERSJ Ltd
References









