


Abstract
α-d-Glucopyranose,3-O-decyl-2-deoxy-6-O-[2-deoxy-3-O-[(3R)-3-methoxydecyl]-6-O-methyl-2-[[(11Z)-1-oxo-11-octadecenyl]amino]-4-O-phosphono-β-d-glucopyranosyl]-2-[(1,3-dioxotetradecyl)amino]-1-(dihydrogen phosphate), tetrasodium salt (E5564) is a second-generation synthetic lipodisaccharide designed to antagonize the toxic effects of endotoxin, a major immunostimulatory component of the outer cell membrane of Gram negative bacteria. In vitro, E5564 dose dependently (nanomolar concentrations) inhibited lipopolysaccharide (LPS)-mediated activation of primary cultures of human myeloid cells and mouse tissue culture macrophage cell lines as well as human or animal whole blood as measured by production of tumor necrosis factor-α and other cytokines. E5564 also blocked the ability of Gram negative bacteria to stimulate human cytokine production in whole blood. In vivo, E5564 blocked induction of LPS-induced cytokines and LPS or bacterial-induced lethality in primed mice. E5564 was devoid of agonistic activity when tested both in vitro and in vivo and has no antagonistic activity against Gram positive-mediated cellular activation at concentrations up to 1 μM. E5564 blocked LPS-mediated activation of nuclear factor-κB in toll-like receptor 4/MD-2-transfected cells. In a mouse macrophage cell line, activity of E5564 was independent of serum, suggesting that E5564 exerts its activity through the cell surface receptor(s) for LPS, without the need for serum LPS transfer proteins. Similar to (6-O-{2-deoxy-6-O-methyl-4-O-phosphono-3-O-[(R)-3-Z-dodec-5-endoyloxydecl]-2-[3-oxo-tetradecanoylamino]-β-O-phosphono-α-d-glucopyranose tetrasodium salt (E5531), another lipid A-like antagonist, E5564 associates with plasma lipoproteins, causing low concentrations of E5564 to be quantitatively inactivated in a dose- and time-dependent manner. However, compared with E5531, E5564 is a more potent inhibitor of cytokine generation, and higher doses retain activity for durations likely sufficient to permit clinical application. These results indicate that E5564 is a potent antagonist of LPS and lacks agonistic activity in human and animal model systems, making it a potentially effective therapeutic agent for treatment of disease states caused by endotoxin.
The innate immune response has been described as being composed of inherent abilities to respond to both Gram positive and Gram negative bacteria, as well as fungi and other pathogens. Response to Gram negative bacteria is driven, at least in part, by recognizing and responding to endotoxin (lipopolysaccharide or LPS), a major constituent of the outer membrane of Gram negative bacteria. LPS is recognized by divergent pathways. One pathway uses natural antibodies (Reid et al., 1997) and lipoproteins (Wurfel et al., 1995; Wurfel and Wright, 1995; Hailman et al., 1996; Vreugdenhil et al., 2001) to neutralize and clear LPS. A second pathway triggers a vigorous and complex inflammatory response involving cellular activation through lipopolysaccharide binding protein (LBP), cell surface-bound CD14, and toll-like receptor (TLR) 4 (for recent review, see Diks et al., 2001). This latter inflammatory response enables a sensitive and robust reaction to LPS, using it as a “sentinel” molecule, signaling the presence of a potentially infectious agent.
In response to blood-borne infection in mammals, LPS is detected by cells such as monocytes, macrophages, and hepatic Kupffer cells, triggering them to produce a large variety of cytokines, and other cellular mediators (Burrell, 1994; Fiuza and Suffredini, 2001) that can protect the host. However, during and after bacterial killing or when translocated from the lumen of the intestine, LPS unassociated with worsening infection can induce inappropriate (toxic) levels of cellular mediators that trigger pathophysiological events such as hypotension, fever, shock, and coagulopathies (Bone, 1991a,b; Ulevitch and Tobias, 1995; Morrison, 1998; Norimatsu and Morrison, 1998; Suffredini and O'Grady, 1999) often leading to multiorgan failure and death (Brandtzaeg et al., 2001). Intestinal tract-derived endotoxin has been implicated as the cause of a variety of clinical manifestations such as postsurgical inflammatory response after abdominal aortic aneurysm (Roumen et al., 1993; Lau et al., 2000) and coronary artery bypass grafting (Martinez-Pellus et al., 1993), hepatic diseases such as alcoholic cirrhosis (Yin et al., 2001), and aggravation of inflammatory diseases such as graft versus host disease (Cooke et al., 2001) and inflammatory bowel disease (Gardiner et al., 1995).
To prevent LPS toxicity, we have investigated the possibility of developing a receptor antagonist to block its activation of cells. Lipid A is the unique fatty-acylated diphosphorylated diglucosamine portion of LPS that is a common element of LPS from most pathogenic bacteria and is its main toxicophore (Galanos et al., 1985a,b; Takada and Kotani, 1989), making antagonism of the interaction of lipid A with target cells an attractive target for the treatment of sepsis, bacteremia, septic shock, and other indications. To this end, we have designed a series of synthetic analogs of lipid A (Rossignol et al., 1999). Previously, we described the synthesis and activity of E5531, an analog of the lipid A from Rhodobacter capsulatus, as an antagonist of LPS (Christ et al., 1995; Kawata et al., 1995) and showed that its antagonistic action involves the cell surface receptor for LPS previously described as TLR4 (Chow et al., 1999).
Although E5531 demonstrated potent inhibition of LPS when added to blood in vitro and in vivo, activity decreased as a function of time. This reaction has been shown to be due to interaction of E5531 with plasma lipoproteins (Wasan et al., 1999; Rose et al., 2000).
This report describes the activity of E5564, a second-generation LPS antagonist derived from the structure of R. sphaeroides(Rossignol et al., 1999). Compared with E5531, E5564 is structurally and synthetically less complex, yet seems to possess superior activity and pharmacological characteristics. E5564 is an inhibitor of LPS-mediated stimulation of responsive cells in vitro and in vivo as measured by production of cytokines, as well as morbidity and mortality associated with LPS poisoning in animal models.
Materials and Methods
Reagents.
E5564 [α-d-glucopyranose,3-O-decyl-2-deoxy-6-O-[2-deoxy-3-O-[(3R)-3-methoxydecyl]-6-O-methyl-2-[[(11Z)-1-oxo-11-octadecenyl]amino]-4-O-phosphono-β-d-glucopyranosyl]-2-[(1,3-dioxotetradecyl)amino]-1-(dihydrogen phosphate), tetrasodium salt], formula weight 1401.60) was synthesized by Eisai Research Institute of Boston (Andover, MA). E5564 was dissolved at 6.7 mg/ml in sterile 0.01 N NaOH, sonicated for 3 min with an ultrasonicator (VW-380; Misonix, Inc., Farmingdale, NY) then diluted to 100 μM in lactose-phosphate buffer containing (per milliliter) 0.45 mg Na2HPO4 · 7H2O, 0.35 mg NaH2PO4 · H2O, and 100 mg of lactose; made up in sterile water. The pH was adjusted to pH 7.8 with 1 N HCl and this buffered solution of drug stored as aliquots at −20°C until use. For use, each aliquot was thawed only once, and serial dilutions were made in Ca2+/Mg2+-free Hanks' balanced salt solution (Invitrogen, Carlsbad, CA). For in vivo use, commercially formulated E5564 was prepared by treatment of E5564 with NaOH as described above, followed by neutralization, addition of phosphate-buffered lactose, and lyophilization. Vials of 1 mg of E5564 were reconstituted with sterile distilled water and diluted with 5% dextrose in water. The following LPS strains were purchased from List Biologicals (Campbell, CA): Escherichia coli (serotype 0111:B4; trichloroacetic acid-extracted), Klebsiella pneumoniae, Pseudomonas aeruginosa, Salmonella minnesota (wild type), Salmonella typhimurium,Serratia marcescens, and Salmonella minnesotaR595. Salmonella entertidis LPS was purchased from Sigma-Aldrich (St. Louis, MO). E. coli lipid A [serotype 0111:B4, LA-15-PP(506)] was purchased from Daiichi Chemical (Tokyo, Japan). Whole bacteria Enterobacter aerogenes lot E25081, a clinical isolate, and lot ATCC13048 (American Type Culture Collection, Manassas, VA) were grown to late exponential phase in Mueller-Hinton broth, and E. coli were grown overnight at 37°C in heart-brain infusion media (Difco, Detroit, MI), harvested, washed by centrifugation in Dulbecco's phosphate-buffered saline (Ca2+/Mg2+-free), resuspended in distilled deionized water, and lyophilized. Each purified strain of LPS or lyophilized bacteria was solubilized in sterile water for injection and stored as aliquots (21 mg/ml) [either as purified LPS or 1 mg/ml (dry weight) whole bacteria], at −80°C. For use, thawed samples were sonicated for 1 to 2 min as described above immediately before each experiment. Live E. coli(E01292) or S. aureus (E31290) were similarly grown and prepared and used without freezing.
NF-κB Reporter Activity in TLR4-Expressing Cells.
HEK293 cells stably carrying plasmids for TLR4, MD-2, and endothelial leucocyte adhesion molecule-1 (ELAM-1)-luciferase were generated as described previously (Yang et al., 2000) and shown to be responsive to LPS plus CD14 (Hawkins et al., 2002). Cells were seeded in 96-well plates at a density of 20,000 cells/well and maintained in Dulbecco's modified Eagle's medium plus 10% fetal bovine serum for 24 h. The next day, cells were incubated with the indicated concentration of E5564 in the presence of LPS (100 ng/ml) and soluble CD14 (10 nM) for 18 to 20 h. Steady-Glo reagent (Promega, Madison, WI) was added to the wells and the amount of luciferase activity in each sample was quantified in a 1450 MicroBetaTrilux counter (PerkinElmer Wallac, Gaithersburg, MD).
Preparation of Human Whole Blood and Cytokine Assays.
Induction of TNF-α in human whole blood has been described previously (Rose et al., 1995, 2000). Briefly, the concentrations of antagonists indicated in the text and figures were added as 10× stocks in 50 μl of 5% dextrose in water followed by 50 μl of LPS (10 ng/ml final concentration) to 400 μl of heparinized whole blood obtained from normal volunteers (18–51 years old; 50–105 kg) for a total of 500 μl/well (final concentration of whole blood was 80%). After 3-, 4-, 6-, 9-, or 24-h (as indicated) incubation with gentle shaking at 37°C in a 5% CO2 atmosphere, plates were centrifuged at 1000g for 10 min at 4°C and then plasma was drawn off and frozen at −80°C. Plasma was appropriately diluted and tested for TNF-α or interleukins-1β, IL-6, IL-8, and IL-10 and using the appropriate human Predicta ELISA kit (Genzyme Diagnostics, Cambridge, MA).
Assays to measure inactivation of antagonists in whole blood were done essentially as described above, but parallel samples containing the appropriate dilutions of test compound were incubated at 37°C with shaking for 0, 3, or 6 h before adding LPS and then incubated with LPS for 3 h and plasma harvested and assayed for TNF-α as described above.
Animal Care and Handling.
All animals used for harvest of in vitro tissue samples were housed and cared for according to the Guidelines For Care and Use of Laboratory Animals (USDA National Institutes of Health Publication 86-23). Sprague-Dawley male rats were purchased from Charles River Laboratories, Inc. (Wilmington, MA) and 18- to 22-g C57BL/6J male mice from Taconic Farms (Germantown, NY). Three- to 10-week-old Hartley White male guinea pigs were purchased from Elm Hill Breeding Laboratories (Chelmsford, MA). The animal room was maintained at 65 ± 3°F, 45 ± 5% relative humidity with a 12-h light/dark cycle. Animals were fed solid food and tap water ad libitum.
All in vivo experiments were approved by Animal Care and Use Committee of Eisai Co. Ltd. Eight- to 12-week-old C57BL/6 male mice (Japan SLC, Inc., Shizuoka, Japan), 4- to 6-week-old Hartley guinea pigs (Charles River Japan, Inc., Kanagawa, Japan), and 5- to 7-week-old Fischer rats (Japan SLC, Inc.) were housed and cared for in our laboratories. The animal room was maintained at 23 ± 1°C, 45 ± 5% relative humidity with a 12-h light/dark cycle. Animals were fed Agway ProLab (Agway, Inc., Syracuse, NY) as solid food and tap water was given ad libitum. To increase sensitivity of LPS, animals were primed 10 to 12 days before utilization by intravenous injection with 1 to 2 mg/animal of bacillus Calmette-Guerin (BCG; Japan BCG, Inc., Tokyo, Japan) suspended in pyrogen-free saline.
Murine Whole Blood Assays.
Heparinized whole blood was obtained from 8- to 10-week old Sprague-Dawley male rats or 18- to 22-g C57BL/6J male mice. Mice were “primed” by injecting i.v. with an attenuated, live preparation of BCG (2 mg/0.2 ml/tail vein). Blood was collected 10 to 12 days after the injection of BCG from CO2-euthanized animals by using cardiac puncture into syringes containing sodium heparin (LyphoMed Inc., Rosemont, IL) and pooled and stored on ice. One hundred sixty microliters of blood was transferred to wells of a 96-well plate, followed by 20 μl of E5564 and either 20 μl of Hanks' balanced salt solution or LPS. The tissue culture plate was then incubated at 37°C, 5% CO2 for 2 h, on a rotating mixer. Samples were centrifuged (900g, 10 min, 4°C) and the supernatants frozen for subsequent assay for TNF-α or IL-6 by ELISA. Mouse TNF-α and IL-6 were assayed using ELISA mini-kits (Pierce Endogen, Rockford, IL), TNF-α was measured in rat plasma samples by ELISA using a rat TNF-α (BioSource International, Camarillo, CA), and IL-6 was measured in rat plasma samples by a proliferative assay using IL-6-dependent B9 cells (LeMay et al., 1990).
Preparation of Peritoneal Macrophages and Incubations with LPS and E5564.
Peritoneal macrophages were isolated from rats, mice, and guinea pigs treated with 2 mg of a cell wall preparation fromMycobacterium bovis (BCG; Ribi Immunochem Research Inc., Hamilton, MT) as described previously (Kobayashi et al., 1998). Adherent cells were treated with 10 ng/ml E. coli LPS and the indicated amount of E5564 was added to cultures of rat peritoneal macrophages to achieve the concentrations indicated. After a 3-h incubation, plates were centrifuged, and the resulting supernatant samples were stored at −80°C until the cytokine assays were performed. Murine IL-6 and TNF-α was measured as described for plasma (see above). TNF-α in guinea pig peritoneal macrophage cultures was quantified by cytotoxicity (LeMay et al., 1990), whereas IL-6 was measured by bioassay as described above.
Statistical Analysis.
Unless noted, in vitro experiments were done three times using triplicate determinations in each experiment. Mean and S.E. were calculated using standard calculations available in the Microsoft Excel spreadsheet. The E5564 concentration that inhibited 50% of the induced production of cytokine (the 50% inhibitory concentration; IC50) was calculated by a log-linear interpolation between the two points that span the 50% value. For in vivo studies, statistical analyses between the control groups and groups treated with E5564 were performed by one-way analysis of variance or the Fisher's exact test followed by Tukey's multiple comparison test or Dunnett's multiple comparison test. A value of 5% (two-sided) was considered statistically significant.
Results
E5564 Inhibition of LPS-Induced Cytokines in Human Monocytes and Blood.
Assays measuring inhibition of TNF-α induced by 10 ng/ml LPS in adherent human monocytes in the presence of 10% human serum (Christ et al., 1995) indicated that the resultant IC50 value for E5564 in this system was 0.36 ± 0.2 nM.
Similarly, freshly drawn heparinized human whole blood dose dependently responds to LPS by producing TNF-α and other cytokines. As in other model systems, 10 ng/ml LPS generated a near-maximal response for TNF-α that peaked at approximately 3 h. As shown in Fig.1, response was inhibited 100% by 10 nM E5564 with an IC50 value of approximately 1 nM. In three assays the mean IC50 value for inhibition of this response was 1.6 ± 0.3 nM (Table1).

Inhibition of LPS-induced cytokines in human blood. Top, 10 ng/mL LPS plus the indicated concentration of E5564 were added to human whole blood, incubated for 3 h at 37°C, and TNF-α assayed as described under Materials and Methods. Each point and value in the figure represents the mean and standard error of triplicate determinations obtained in a single experiment. No measurable TNF-α was observed in samples incubated without LPS. Statistical significance of inhibition (compared with the LPS-only response) was ∗, p < 0.05 or ∗∗,p < 0.001 by Student's t test. Bottom, 10 ng/ml LPS alone (closed symbols) or plus 10 nM E5564 (open symbols) was added to human whole blood and incubated for 4, 6, 9, or 24 h at 37°C. After the incubation, the blood was centrifuged and the resulting plasma supernatant samples were assayed for TNF-α (●, ○), IL-1β (♦, ⋄), IL-6 (▾, ▿), IL-8 (▴, ▵), or IL-10 (▪, ■). Complete inhibition of release of all cytokines was observed in the presence of 100 nM E5564 (data not shown). Incubation of unstimulated blood samples for up to 24 h induced less than 1% of peak levels for TNF-α, IL-1β, IL-6, and IL-10 and less than 4% of peak levels of IL-8 from samples containing LPS. Each point and value in the Figure represents the mean and standard error of triplicate determinations. This experiment was performed three times with similar results. Statistical significance of inhibition (compared with the LPS-only samples) for the three assays wasp < 0.05 for all values of IL-8 and IL-6 measured in the presence of 10 nM E5564 and p < 0.001 for all values of IL-10, IL-1β, and TNF-α (except for the 24-h TNF-α time point) measured in the presence of 10 nM E5564 (Student'st test).
E5564 antagonism of LPS- induced cytokine generation in human blood
Inhibition of induction of other cytokines was also tested in whole blood over incubation periods of 4, 6, 9, or 24 h. As described in Fig. 1, IL-6 levels remained elevated throughout the 24-h incubation period, and over the same 24-h incubation period, levels of IL-1β rose to 1,077.9 ± 181.4 pg/ml, IL-8 to over 20,000 pg/ml, and IL-10 to over 450 pg/ml.
E5564 (10 nM) inhibited TNF-α and IL-1β production by 100% at all times tested. Similarly, LPS-induced IL-6 production was inhibited greater than 94% by 100 nM E5564, with a mean IC50 value of less than 2 nM (Table 1). Complete inhibition of IL-8 production required slightly higher E5564 concentrations. LPS-induced IL-8 production was inhibited >70 to 90% by 100 nM E5564 with an IC50 value of 13 nM over the 24-h incubation time. IL-10 production was inhibited 100% by 10 nM E5564, and the resulting IC50 of <1 nM E5564 against this cytokine (Table 1).
Potency of E5564 was dose-dependent for agonist. Other in vitro assays in whole blood, indicated that if LPS concentration was reduced 10-fold, the IC50 value of E5564 was reduced approximately 2- to 4-fold (data not shown).
The possibility that E5564 demonstrated agonistic activity in human blood was tested by addition of 10 μM E5564 alone for up to 9 h. In all experiments, resultant TNF-α and/or IL-6 levels with were at or below basal values, indicating that E5564 possesses no LPS-like agonistic activity.
Antagonistic Effects of E5564 on TNF-α Production Induced by LPS from Different Strains of Bacteria, Whole Bacteria, and E. coli Lipid A.
Lipopolysaccharides derived from various strains of Gram negative bacteria induced TNF-α in human blood. These LPSs demonstrated different dose dependencies, so for comparison purposes, their concentrations were adjusted to stimulate approximately similar amounts of TNF-α to that induced by the standard E. coli LPS (strain 0111:B4) at 10 ng/ml (Table2).
E5564 inhibition of TNF-α induced by LPS from various strains of bacteria, dead bacteria, and lipid A
The IC50 values (mean ± S.E.) for antagonism of LPS response by E5564 are also shown in Table 2. For all strains of LPS the IC50 value for E5564 was between 1.0 and 12.4 nM, indicating that E5564 is a potent antagonist of these different strains of LPS.
As observed with purified LPS, whole E. coli or E. aerogenes bacteria induced TNF-α (Table 2). E5564 similarly antagonized this activation with IC50 values of 0.65 to 1.5 nM, indicating that E5564 can potently block activation by whole Gram negative bacteria.
TNF-α induced by 10 ng/ml lipid A was inhibited 77% by 10 nM E5564 and 100% by 100 nM E5564, with an average IC50value of 1.2 ± 0.7 nM (Table 2).
Antagonism of Cellular Activation by Bacteria in Human Blood.
To determine whether cellular activation by live bacteria is inhibited by E5564, antagonism of live Gram negative bacteria (E. coli) and live Gram positive bacteria (S. aureus) was tested. As shown in Fig. 2, E5564 inhibited induction of TNF-α by high doses of Gram negative bacteria, but was inactive against Gram positive (S. aureus) bacteria at concentrations up to 1 μM. These results indicate that E5564 is active against cellular activation by Gram negative bacteria, but not Gram positive bacteria (i.e., activation through TLR2).

Effects of E5564 on induction of TNF-α by Gram negative and Gram positive bacteria. The indicated concentration of E5564 was added to heparinized human blood immediately before inoculation with either 1 × 105 cfu/ml E. coli (E01292; ●) or 1 × 106 cfu/mlS. aureus (E31290; ●, ▪), incubated for 3 h, and assayed for plasma TNF-α as described under Materials and Methods. Each point and vertical line represent mean and S.E.M. of 4 to 12 experiments.
Time-Dependent Inhibition of Antagonistic Activity by Serum.
Side-by-side comparisons of E5564 to E5531 (our “first-generation” antagonist) indicated that E5564 is nearly 7-fold more potent an inhibitor of TNF-α production than E5531 [IC50value for E5564 = 1.5 ± 0.37 versus 10.4 ± 3.1 nM for E5531 (n = 7 assays)].
The effects of interaction of E5564 and E5531 with blood components was evaluated by preincubating a wide range of concentrations of antagonists with whole blood for 0, 3, or 6 h before addition of LPS. As shown in Fig. 3, the apparent IC50 value of E5564 increased with time of preincubation in blood (apparent IC50 = 1.5 ± 0.4 nM at zero time, 5.9 ± 1.8 nM at 3 h, and 7.7 ± 2.5 nM after 6-h preincubation in whole blood). Corresponding IC50 values for E5531 were 10.4 ± 3.1 nM at zero time, 28.8 ± 8.8 nM at 3 h, and 50.5 ± 16 nM after 6-h preincubation. These results indicate that low concentrations of E5564 maintain their antagonistic activity longer than similar concentrations of E5531. Higher doses of E5564 (e.g., 1 μm) completely inhibited response throughout the entire 6-h incubation period. These results indicate that even though E5564 is inactivated by blood, it is likely that dosing can be adjusted to maintain effective levels of antagonistic activity over time.
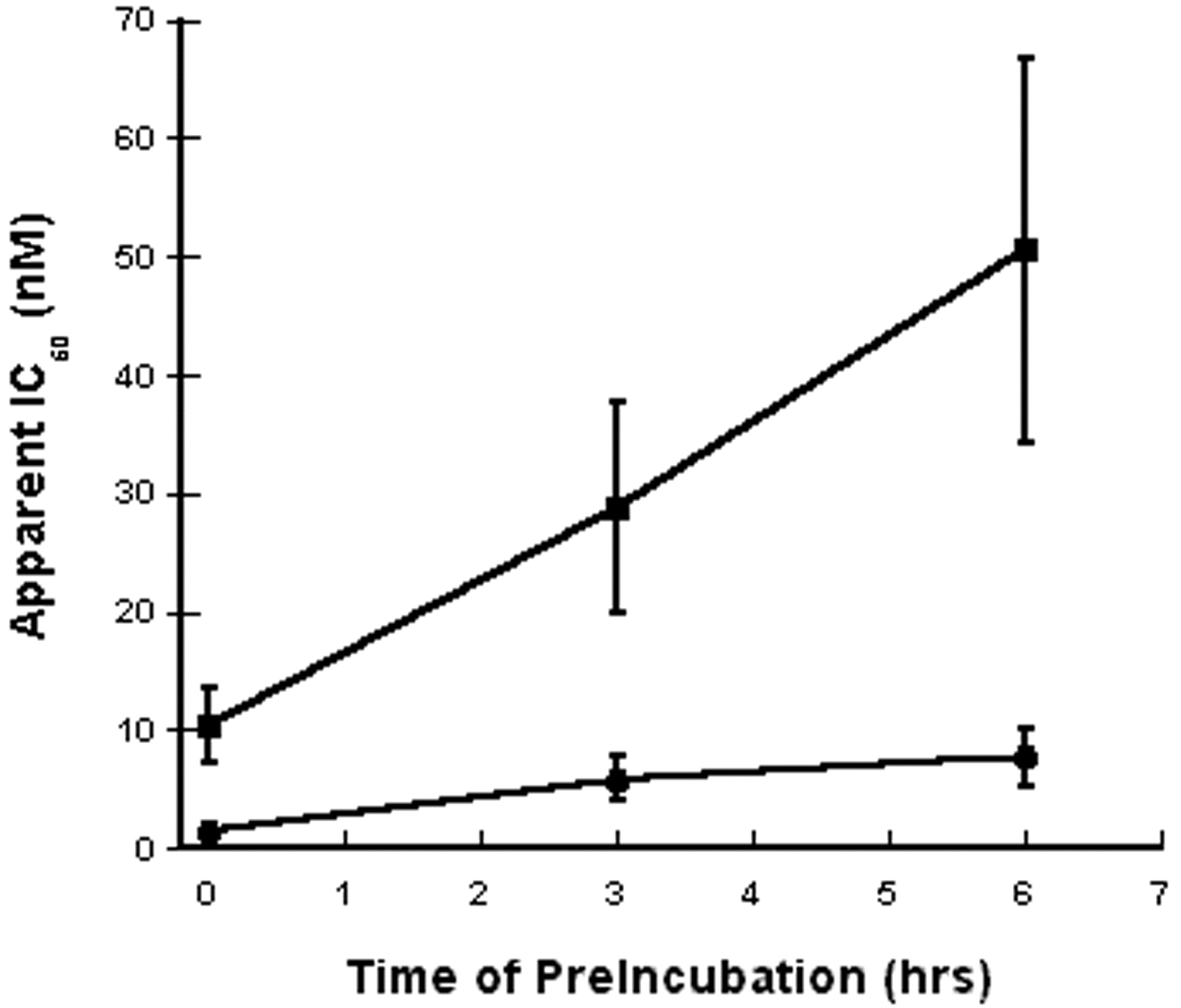
Loss of antagonistic activity during incubation in whole blood. Tenfold dilutions of E5564 (●) or E5531 (▪) were preincubated in human whole blood for the indicated time (0, 3, or 6 h) at 37°C, and 10 ng/ml LPS (final concentration) was added and the incubations continued for an additional 3 h. Plasma TNF-α was measured, and IC50 values were interpolated as described under Materials and Methods. The differences in IC50 values between E5564 and E5531 were significant (p < 0.05) at each time point (Student'st test).
Antagonistic Activity of E5564 in Murine and Guinea Pig Macrophages and Blood.
Mouse peritoneal macrophages incubated with 10 ng/mlE. coli endotoxin for 2 h released 3315 ± 318 pg/ml TNF-α and 5.0 ± 0.53 ng/ml IL-6. In this assay, E5564 at a concentration of 100 nM inhibited release of TNF-α by 95% and IL-6 by 89%. The IC50 value for E5564 was 20.4 ± 12.5 nM against TNF-α and 16.6 ± 6.7 nM against IL-6 (Table3).
E5564 inhibition of TNF-α and/or IL-6 induced by LPS in peritoneal macrophages and whole blood from mice, rats, and guinea pigs
In mouse whole blood, survey studies have previously indicated that ex vivo induction of TNF-α was not robust or reproducible. However, IL-6 was found to be dose dependently stimulated by LPS with maximal response at 10 μg/ml LPS, and greatest dose dependence at 1 to 100 ng/ml LPS. This response was dependent on time of incubation with LPS for only up to 2 h with little or no further increase seen thereafter (data not shown). After 2-h incubation with 10 ng/ml LPS, IL-6 concentrations rose to 13 ± 0.18 ng/ml (n = 3) compared with 0.158 ± 0.013 ng/ml in samples that were not treated with LPS. E5564 at a concentration of 100 nM inhibited the IL-6 increases by 85%, and the IC50 value for E5564 was 20.2 ± 7.0 nM in these combined experiments (Table 3).
To test for inhibition of nitric oxide production, cultured mouse macrophages (RAW 264.7) were incubated overnight with 10 ng/ml LPS, which induced accumulation of more than 20 μM nitrite in the culture medium. E5564 dose dependently inhibited this induction [IC50 = 91 ± 36 nM (mean ± S.E.;n = 5)] with >95% inhibition observed at 1 μM E5564 (data not shown).
Rat peritoneal macrophages stimulated with 10 ng/ml LPS induced 2867 ± 326 pg/ml TNF-α, whereas induction of IL-6 was robust but variable with release of between 23 and 163 ng of IL-6 per milliliter of culture medium. E5564 at 100 nM inhibited TNF-α and IL-6 production by 89%. The IC50 value for E5564 was 7 ± 5.6 nM for TNF-α and 16.2 ± 17.5 nM (28.6 and 3.9 nM) for IL-6 (Table 3).
E5564 was significantly less active in rat blood than in rat peritoneal macrophages. At a concentration of 1000 nM, E5564 inhibited LPS-induced TNF-α increases by 86%, and the IC50 for E5564 was 136 ± 61 nM in these combined experiments. In the same incubations where TNF-α was measured, 10 μM E5564 inhibited the LPS-induced IL-6 production by 70% with an average IC50 value of ∼2400 nM.
Guinea pig macrophages incubated with 10 ng/ml E. coli LPS for 3 h released 1897 ± 348 pg/ml TNF-α (n= 3) and 3.0 ± 0.43 ng/ml IL-6 (n = 2). E5564 (10 nM) inhibited LPS-induced TNF-α and IL-6 by >98%, with a resultant IC50 value for E5564 of 0.30 ± 0.15 nM for TNF-α and 0.5 ± 0.3 nM for IL-6 (Table 3).
Inhibition of LPS Induced TNF-α Release in Vivo.
E5564 or vehicle was injected intravenously into BCG-primed mice along with 100 μg/kg LPS, a lethal dose. One hour after administration, blood was collected and TNF-α levels measured (Fig.4). E5564 administered at 30, 100, 300, or 1000 μg/kg suppressed plasma TNF-α concentrations by 24, 38, 81, and 93%, respectively.

Effects of E5564 on TNF production in BCG-primed animals. The indicated dose of E5564 or vehicle was injected intravenously into mice, guinea pigs, and rats (n = 5) along with 100, 1000, and 3 μg/kg LPS, respectively. One hour after administration, blood was collected and TNF-α levels measured. ∗, p < 0.05 versus control group by one-way analysis of variance followed by Dunnett's multiple comparison.
BCG-primed guinea pigs were intravenously injected with a lethal dose of LPS (1000 μg/kg) and E5564 or vehicle and blood was collected 1 h later for measurement of TNF-α levels (Fig. 4). Administration of 10, 30, 100, and 300 μg/kg E5564 suppressed LPS-induced plasma TNF-α concentrations by 29, 57, and 94%, respectively. The ED50 value for this model system was estimated to be 37 μg/kg E5564.
Similarly, BCG-primed rats were intravenously administered E5564 or vehicle along with 3 μg/kg LPS (a nonlethal dose), and blood was collected 1 h later for assay of TNF concentrations (Fig. 4). Administration of E5564 inhibited induction of TNF-α by 84, 97, and 100%, by 10, 100, and 1000 μg of E5564/kg, respectively.
Effect of E5564 on LPS-Induced Lethality in Mice.
To evaluate the ability of E5564 to prevent LPS-induced mortality, BCG-primed mice were injected intravenously with 100 μg/kg LPS along with the indicated doses of E5564 or vehicle, and incidence of mortality was monitored for 72 h (Fig. 5). Whereas the administration of 100 μg/kg LPS alone resulted in 90% mortality by 72 h, coinjection of E5564 significantly and dose dependently reduced the incidence of mortality.
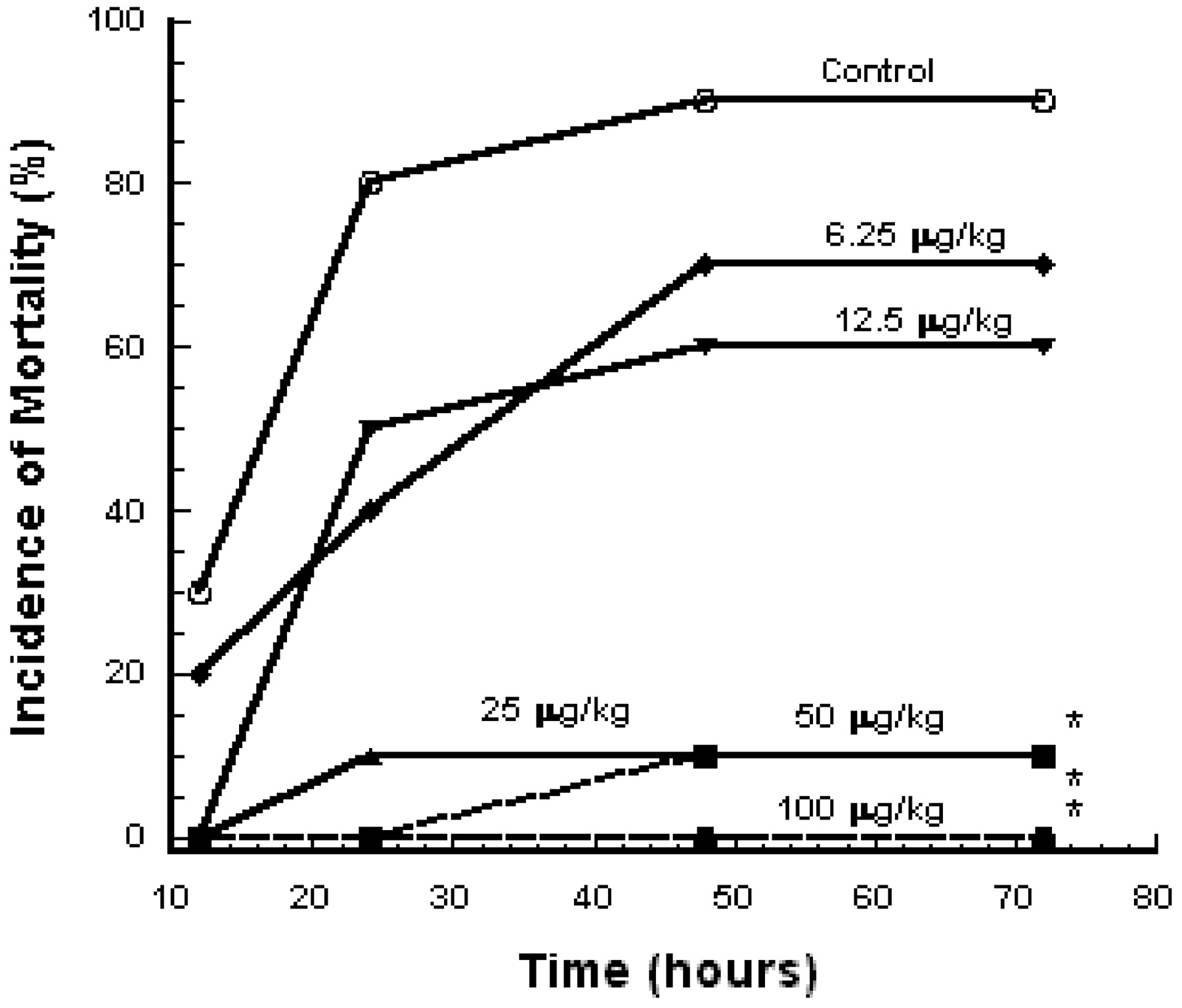
Effects of E5564 on mortality induced by E. coli LPS in BCG-primed mice. Vehicle (○) or E5564 6.25 μg/kg (♦), 12.5 μg/kg (▾), 25 μg/kg (▴), 50 μg/kg (▪), or 100 μg/kg (●) was injected intravenously into mice (n = 10) along with 100 μg/kg LPS. Lethality was observed during 72h. ∗, p < 0.05 versus control group by Dunnett's multiple comparison test. Incidence of mortality was monitored for 72 h.
Effects of E5564 on Septic Shock Caused by Bacterial Infection in Mice.
The objective of the study was to examine the use of E5564 in conjunction with the β-lactam antibiotic latamoxef to prevent mortality in mice injected with E. coli. A suspension ofE. coli (3.23 × 107 cfu/animal) was injected intraperitoneally into BCG-primed mice. One hour later, the mice were injected intravenously with vehicle, E5564 alone (5 mg/kg), latamoxef alone (30 mg/kg), or E5564 and latamoxef together. The incidence of mortality was recorded for 72 h after infection.
As shown in Fig. 6, mortality of theE. coli-infected control group (no treatment) reached 90%. Administration of either latamoxef or E5564 alone resulted in a modest reduction in the incidence of mortality. However, simultaneous administration of E5564 and latamoxef suppressed mortality to 20%. These results demonstrate that combined treatment with E5564 and latamoxef is significantly more effective than treatment with antibiotic alone or E5564 alone in reducing the incidence of mortality caused by E. coli infection in BCG-primed mice.

Effects of E5564 on mortality induced by E. coli infection in BCG-primed mice. A suspension of E. coli (3.23 × 107 cfu/animal) was injected intraperitoneally into BCG-primed mice. One hour after inoculation, vehicle (●), 30 mg/kg latamoxef (♦), or 5 mg/kg E5564 alone (▪) or with latamoxef (▴) was injected intravenously into mice. The incidence of death was recorded at 12, 24, 48, and 72 h after administration. Results are expressed as percentage of mortality of the total number of animals in each group (n = 20). ∗, p < 0.05 versus the other three groups by Fisher's exact test followed by Tukey's multiple comparison test.
Lack of Agonistic Activity of E5564 in Vivo.
E5564 was intravenously injected into BCG-primed animals (3000 μg/kg into mice, 300 μg/kg into guinea pigs, and 100 and 1000 μg/kg into rats) and blood was collected for analysis of TNF-α 1 h later. Neither E5564 nor vehicle alone caused any marked induction of TNF-α, indicating that E5564 is devoid of endotoxin-like activity in this assay.
How Does E5564 Work?
Interaction of E5564 at the TLR4 receptor for LPS has been investigated by measuring response to LPS (expression of NF-κB-driven luciferase) by HEK293 cells made responsive to LPS by transfection with TLR4 and MD-2. As shown in Fig.7, luciferase expression in this system is dependent on LPS plus soluble CD14 and inhibited by E5564 (IC50 = 32 nM). In contrast, E5564 did not inhibit heat-killed S. aureus activation of TLR2-transfected HEK293 cells at concentrations up to 1 μM (J. Chow, unpublished data). These results suggest that E5564 interferes with LPS signaling at the TLR4 receptor and/or soluble CD14.
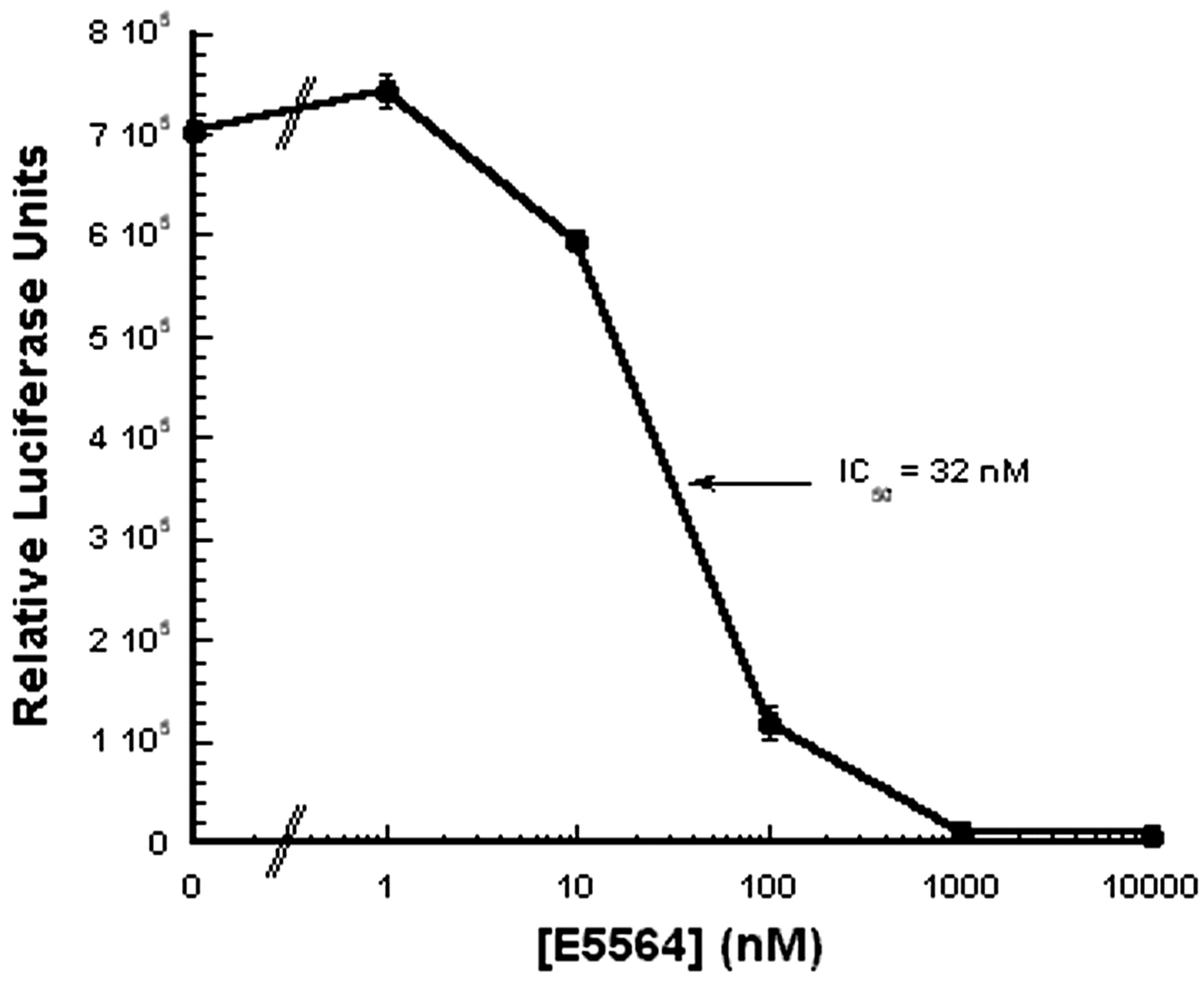
E5564 inhibits LPS-induced TLR4-mediated NF-κB reporter activity. HEK293 cells stably transfected with TLR4, MD-2, and a luciferase reporter gene driven by an NFκB-dependent promoter were incubated with the indicated concentrations of E5564 with 100 ng/ml LPS and 10 nM sCD14 for 18 to 20 h. The next day, luciferase activity was quantified as described under Materials and Methods. This experiment is representative of three and each point represents the mean ± S.D., n = 3.
In addition, because LPS associates with soluble serum receptors such as LBP and sCD14 before interacting with TLR4, these proteins or other soluble serum factors could also be critical “receptor(s)” for E5564 or be necessary for antagonistic activity. To determine whether E5564 needs to be present in the culture media and/or serum or whether pretreatment of the cell surface can block LPS-mediated activation, RAW 264.7 cells were tested for inhibition of TNF-α release after various drug pretreatment regimens. Pilot experiments indicated that treatment of cells with E5564 followed by rapid, extensive cell washing and addition of LPS along with serum or LBP for 3 h resulted in clear dose-dependent inhibition of LPS activity. As shown in Table4, addition of E5564 in the absence of serum followed by washing (three times) and addition of serum plus LPS, resulted in IC50 values of 0.8 ± 0.1 nM (n = 3). If serum is included in this E5564 pretreatment, the resultant IC50 value was unchanged at 1.1 nM (n = 2), indicating that serum did not increase or dramatically decrease the potency of E5564. Similarly, purified LBP used in the place of serum had no effect on E5564 activity (data not shown).
Activity of E5564 after different conditions of preincubation and dissociation incubation with cells
To determine whether E5564 retained its activity after unbound material was removed from the adherent cells or whether it is readily released from the cell surface, cells were treated with different concentrations of E5564 in the absence or presence of serum, followed by washing and incubating for up to 2 h with and without serum to allow antagonist dissociation. LPS plus serum was then added and cellular stimulation measured by TNF-α release. The resulting IC50 values for E5564 were unchanged after 30-min incubation in serum-free medium (0.7 ± 0.1 nM) and increased slightly (2.5 nM) when serum was included in the incubation medium. Extending the “washout” period to 2 h in the presence of serum further increased the IC50 value to 24 to 25 nM, indicating a slow loss of “surface-associated” antagonistic activity.
Discussion
A variety of pathologies have been attributed to responses to endotoxin even in cases where identification of its source is unclear. In the clinic, antibiotic treatment to control bacterial infection has been reported to result in the release of endotoxin and consequent aggravation of septic shock. Although the source of endotoxin during infection by Gram negative bacteria may seem obvious, endotoxemia has also been reported to occur during Gram positive and fungal infection (Opal et al., 1999). In addition, it has been reported that endotoxin is translocated from the intestinal tract to the splanchnic circulation under a variety of conditions. Perhaps more definitive proof that pathological outcomes are truly due to endotoxin awaits the therapeutic application of an effective endotoxin antagonist.
Mechanism of Action of E5564
It is not clear exactly how E5564 works to block LPS signaling. Because E5564 is a structural analog of the lipid A portion of LPS, it is logical to hypothesize that the antagonist interacts with the same signaling components that interact with LPS such as the soluble serum proteins LBP and sCD14, as well as membrane-associated CD14 and perhaps the TLR4/MD-2 receptor complex. In the present study, E5564 blocked LPS/sCD14-induced reporter activity in TLR4/MD-2-expressing HEK293 (Fig. 7), but not TLR2-mediated signaling by heat-killed S. aureus. These findings indicate that E5564 selectively inhibits LPS signaling via TLR4/MD-2. However, a limitation to this model system is that LPS requires the presence of sCD14 for cellular activation, making it difficult to determine whether E5564 blocks LPS binding to sCD14 or TLR4/MD-2. Results from experiments described in Table 4indicated that serum components neither increased nor dramatically decreased the potency of E5564, indicating that they are not critical to E5564 antagonistic activity.
Further support of the hypothesis that interaction of E5564 at CD14 does not play a key role in its activity comes from a previous study byLien et al. (2000) describing the activity of novel synthetic acyclic lipid A-like agonists that activate TLR4/MD-2 in the absence of CD14. E5564 inhibited the actions of these agonists under serum-free conditions. Taken together, these lines of evidence make it tempting to speculate that E5564 binds to TLR4/MD-2 complex, thereby blocking LPS binding or transmembrane signaling.
The downstream effect of inhibiting the initial signaling by LPS seems to be an inhibition of all LPS-induced cytokines measured, including TNF-α, IL-1β, IL-6, IL-8, IL-10, and nitric oxide, which was measured in cultured cells, whole blood, and in vivo.
The likelihood that E5564 does not inhibit the interaction of LPS with soluble receptors may increase its value as a therapeutic agent. Because endotoxemia and sepsis induce acute phase in the liver, plasma levels of proteins such as LBP change dramatically (Opal et al., 1999). Therapy targeted toward these serum proteins would require dosing adjustments to accommodate increased synthesis or turnover of these proteins.
Comparisons of antagonistic potency in cells cultured in 10% serum versus whole blood allow us to determine whether the high concentration of proteins/lipoproteins present in serum inhibit E5564 activity. In all systems but the rat, antagonistic activity of E5564 in cultured cells was within 4-fold that measured in high serum (blood) compared with assays done in low-serum conditions (cultured cells or monocytes). This indicates that serum has little or no inhibitory effect on antagonistic activity under these in vitro conditions. However, extended incubations in whole blood demonstrated that activity of E5564 was measurably reduced. Other studies (K. M. Wasan, O. Sivak, R. A. Cote, A. I. MacInnes, K. D. Boulanger, M. Lynn, W. J. Christ, L. D. Hawkins, and D. P. Rossignol, manuscript in preparation) indicate that like E5531, E5564 is not rapidly metabolized, but binds to lipoproteins, and time dependently loses antagonistic activity. The observation that lipoproteins reduce drug activity may explain the poor activity of E5564 in rat blood that has a relatively high lipoprotein content (Segrest and Albers, 1986).
Is Endotoxin the Major Component from Gram Negative Bacteria That Activates Cytokine Response in Blood?
It has been postulated that bacterial components other than endotoxin may activate cells in whole blood, and recently, different subtypes of toll-like receptors have been implicated in responses to these different components. However, this differentiation of response to different receptors does not indicate the relative importance of these components in cellular stimulation. E5564 seems to be a specific antagonist for the TLR4 receptor and is inactive against TLR2-directed agonists (Heine et al., 2000). Based on the ability of E5564 to inhibit cellular activation by LPS from a variety of Gram negative bacteria, whole killed or live bacteria, but not Gram positive bacteria, it is likely that E5564 is an effective antagonist for activation of blood cells specifically by Gram negative bacteria. Furthermore, the observation that E5564 is a potent and complete (or nearly complete) inhibitor of Gram negative bacteria in the bacterial sepsis model helps clarify the role of endotoxin in activation of cells in blood. Effective reduction of inflammatory response and death due to bacterial infection by E5564 indicates that endotoxin or other agents that bind to TLR4 drive response to Gram negative bacteria.
Therapeutic Potential of E5564 as an Endotoxin Antagonist
E5564 Is Being Developed as an Endotoxin Antagonist for Human Therapeutic Use.
Safe treatment with E5564 requires that it generate no LPS-like response on its own (Rossignol et al., 1999). When added alone to whole blood or injected into BCG-primed animals, E5564 was devoid of agonistic activity (cytokine generation or overt physiological effects) at concentrations as high as 100 μM in vitro or up to 1000 μg/animal (the highest concentrations and doses tested).
During extended incubation in whole blood, E5564 retained activity better than similar concentrations of the first-generation antagonist E5531. Based on the proposed mechanism of action as a cell surface antagonist, it is likely that E5564 can completely block cellular activation by LPS. This block is achieved by concentrations of E5564 as low as 10 nM (14 ng/ml) in vitro, and at doses of 1 mg/kg or less in animal models challenged with lethal doses of endotoxin. In animals lethally infected with E. coli, treatment of the infection with antibiotic alone did not prevent death in a majority of the animals; however, combining antibiotic with 5 mg/kg E5564 reduced mortality to one-third that of antibiotic alone. It is important to note that in this model, infection and endotoxin exposure began 1 h before administration of antiendotoxin therapy, differing from our prophylactic treatment models in that endotoxin is present before administration of E5564 and representing a situation more like that of human infection. We have previously shown that this model involves profound endotoxemia, especially after antibiotic treatment (Christ et al., 1995; Kobayashi et al., 1998); however, we have not measured plasma endotoxin levels in treated animals in this study because E5564 is a potent activator of the limulus assay, likely due to its structural similarities to Lipid A (data not shown).
Both our LPS-challenge model and infection model use animals that have been sensitized or primed to LPS by previous infection with BCG, increasing cytokine response and lowering the threshold lethal dose of endotoxin. All animal models of sepsis and infection have been criticized for their inability to closely mimic human sepsis. However, we believe that the primed model is the most relevant to the study of endotoxin antagonists such as E5564. It is well known that compared with humans, unprimed rodents such as rats and mice and primates demonstrate a profound insensitivity to endotoxin, requiring endotoxin doses as high as milligrams per kilogram, whereas humans demonstrate reproducible response to endotoxin at doses as low as 2 ng/kg. This argues that either LPS contributes only weakly to the inflammatory process in animal models, or that response to infection occurs only after the level of infection is very high, representing a process different from that in more LPS-sensitive species such as humans.
Even in primed animal models, lethal doses of LPS in are high, approximately 100 μg/kg, generating estimated plasma concentrations of ∼1 μg/ml. These plasma levels are still >100-fold that found in even the most extreme cases of human sepsis (Opal et al., 1999). Because the dose of E5564 required to protect against LPS is proportional to the LPS challenge dose, studying E5564 in these animal models indicates that E5564 can be a safe and effective antagonist even under these extraordinary conditions. E5564 is approximately 10-fold better in human blood than mouse blood (IC50 = 1.6 nM in human whole blood; Table 2 versus ∼20 nM in mouse whole blood; Table 3). Taken together, comparison of in vitro and in vivo results leads us to believe that in clinical use, we may be able to readily establish plasma concentrations of E5564 that will provide us with a wide margin of efficacy against even very high plasma concentrations of LPS.
Complete block of cytokine response by 10 nM E5564 in blood extrapolates to a human dose of ∼100 μg in a 70-kg individual. Recent studies have supported this extrapolation by finding that a dose of 100 μg of E5564 given to normal volunteers over 30 min completely blocks response (signs, symptoms, and cytokines) to a dose of 4 ng/kg endotoxin administered at the midpoint of the E5564 infusion (Lynn et al., 2003).
In vitro and ex vivo assays have found that low concentrations of E5564 time dependently lose ability to inhibit LPS response. In light of these observations, it is perhaps not surprising that low doses of E5564 demonstrate a time-dependent loss of activity after administration into normal volunteers. This loss in activity is overcome when E5564 doses are increased (D. P. Rossignol, K. M. Wasan, N. Wong, L. Q. Ngyen, J. Rose, J. Moran, and M. Lynn, manuscript in preparation). Phase I clinical safety and tolerability assays indicate that E5564 is safe and except for the occurrence of phlebitis, well tolerated at doses up to 252 mg administered over 72 h. At this dose, in vivo antagonistic activity is retained for at least an additional 72 h after discontinuing infusion. This leads us to believe that sufficient therapeutic activity can readily be administered to patients.
In conclusion, we have found that E5564 is a highly active antagonist of LPS in vitro in human and animal systems. This activity translates into effective in vivo antagonism of endotoxin in animal models, resulting in enhancement of survival after challenge with endotoxin or bacterial infection. This consistent, potent activity combined with a clear lack of “LPS-like” agonistic activity leads us to believe that E5564 can be a safe clinical therapeutic for the treatment of endotoxemia, including sepsis and septic shock.
Acknowledgments
We thank Donna Young for excellent technical assistance. Whole bacteria Enterobacter aerogenes was obtained from Dr. Naoaki Watanabe (Tsukuba Research Laboratories). Lot ATCC13048 was obtained from American Type Culture Collection (Manassas, VA), and Lot E25081was a clinical isolate.
Footnotes
-
↵1 Current address: Harvard Medical School, Department of Membrane Transport, Boston, MA 02115.
-
↵2 Current address: Eisai Medical Research Inc., Glenpointe Centre W., 500 Frank W. Burr Blvd., Teaneck, NJ 07666-6741.
-
DOI: 10.1124/jpet.102.044487
- Abbreviations:
- LPS
- lipopolysaccharide
- LBP
- lipopolysaccharide binding protein
- TLR
- toll-like receptor
- NF-κB
- nuclear factor-κB
- HEK
- human embryonic kidney
- TNF-α
- tumor necrosis factor-α
- IL
- interleukin
- ELISA
- enzyme-linked immunosorbent assay
- BCG
- bacillus Calmette-Guerin
- cfu
- colony-forming unit
- E5531
- (6-O-{2-deoxy-6-O-methyl-4-O-phosphono-3-O-[(R)-3-Z-dodec-5-enoyloxydecyl]-2-[3-oxo-tetradecanoylamido]-β-O-phosphono-α-d-glucopyranose tetrasodium salt
- Received September 17, 2002.
- Accepted November 15, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}