


Abstract
The extent to which the prostacyclin (IP) receptor regulates the release of two proinflammatory chemokines from human airway epithelial cells was investigated using the novel and competitive IP-receptor antagonist 4,5-dihydro-1H-imidazol-2-yl)-[4-(4-isopropoxy-benzyl)-phenyl]-amine (RO1138452). In BEAS-2B human airway epithelial cells, taprostene, a selective IP-receptor agonist, suppressed interferon-γ-induced CXCL9 and CXCL10 release in a concentration-dependent manner. These effects were mimicked by 8-bromo-cAMP, and they were abolished in cells infected with an adenovirus vector encoding a highly selective inhibitor of cAMP-dependent protein kinase (PKA). RO1138452 blocked the inhibitory effect of taprostene on chemokine output in a manner inconsistent with surmountable competitive antagonism. Comparable results were obtained using primary cultures of human airway epithelial cells. The basis of the antagonism imposed by RO1138452 was studied further using BEAS-2B cells stably transfected with a cAMP-response element (CRE) luciferase reporter. On this output, RO1138452 also behaved insurmountably. Mechanistically, this could not be attributed to covalent receptor inactivation, allosterism, or a state of hemiequilibrium. Other studies established that the degree by which RO1138452 antagonized taprostene-induced CRE-dependent transcription was not reversed over a 20-h “washout” period. This pharmacological profile is consistent with the behavior of a pseudo-irreversible antagonist where dissociation from its cognate receptor is so slow that re-equilibration is not achieved at the time the response is measured. Collectively, these data provide compelling evidence that human airway epithelial cells express inhibitory IP-receptors linked to the activation of PKA. Moreover, contrary to existing literature, RO1138452 behaved pseudoirreversibly, emphasizing the need, in drug discovery, to screen potential new medicines in the target tissue(s) of interest.
Prostacyclin (PGI2) is a labile eicosanoid produced by the wall of arterial blood vessels (Bunting et al., 1976), and it is derived from arachidonic acid following the sequential action of cyclooxygenase and PGI2 synthase (Vane and Botting, 1995). Like other prostanoids formed by the cyclo-oxygenation of arachidonic acid, the biological actions of PGI2 are mediated through one or more G protein-coupled receptors (GPCRs). Studies performed since the early 1980s have provided pharmacological evidence for five main classes of GPCRs for the naturally occurring prostanoid agonists, and these classes have been designated DP, EP, FPGFα receptor, IP, and TP (Narumiya et al., 1999). This taxonomy was formulated from rank orders of agonist potency obtained in various pharmacological preparations where the first letter denotes the agonist most selective at that receptor, with this being at least 1 order of magnitude more potent than the other natural ligands. Molecular techniques have now confirmed this pharmacological classification with the cloning and expression of the five prostanoid receptors in a number of species, including the human (Narumiya et al., 1999).
Currently, the explicit classification of responses mediated by IP-receptors is hindered by a paucity of suitable pharmacological tools. Indeed, PGI2 and many PGI2 analogs and mimetics, including iloprost, carbacyclin, AFP-07, and TEI-9063 (see Wise and Jones for details), are not sufficiently selective for their biological actions to be diagnostic of IP-receptor agonism. Even cicaprost, which is often the agonist of choice in studies examining IP-receptor pharmacology, must be used with caution because it does not effectively discriminate IP-, EP4-, and, to a lesser extent, EP3-receptor-mediated responses (Abramovitz et al., 2000; Wise and Jones, 2000). To overcome these problems, we have used here a synthetic PGI2 analog, taprostene (Schneider et al., 1993). This ligand was initially described as a full agonist (Jones et al., 1997). However, subsequent studies established that it has relatively lower efficacy compared with other IP-agonists and that it behaves as a partial agonist in tissues where IP-receptor density is limiting and/or receptor-effector coupling efficiency is low (Wise and Jones, 2000; Jones and Chan, 2001; Chan and Jones, 2004; Chow et al., 2004). Nevertheless, taprostene is selective for the IP-receptor subtype across several species, including the human (Chan and Jones, 2004; Chow et al., 2004) (Supplemental Fig. 1), and it was the ligand of choice for most experiments described herein. Moreover, several IP-receptor antagonists based on the 2-(phenylamino)imidazoline template were reported recently (Clark et al., 2004; Bley et al., 2006). Some of these compounds, including RO1138452 (Fig. 1), are highly selective for the IP-receptor subtype, and they act competitively, with affinities in the low nanomolar range (Jones et al., 2006). Thus, these pharmacological tools permit the (patho) physiological role of IP-receptors to be evaluated unambiguously that, hitherto, has not previously been possible.
Accumulating evidence suggests that agonism of IP-receptors in the lung may exert anti-inflammatory activity, antiviral activity, or both (Jaffar et al., 2002; Takahashi et al., 2002; Hashimoto et al., 2004; Zhou et al., 2007a,b). For example, exposure of sensitized IP-receptor-deficient mice to allergen is associated with enhanced pulmonary inflammation compared with wild-type animals (Takahashi et al., 2002). Similarly, agonism of IP-receptors has been shown to stimulate the production of interleukin-10 from murine CD4+ T-helper 2 cells, and, in vivo, to attenuate allergen-induced, T-helper 2 cell-mediated inflammation (Jaffar et al., 2002). Given the emerging anti-inflammatory activity of PGI2 in the lung, two principle objectives formed the basis of the research described herein. First, we wanted to extend the experiments performed with mice (described above) to a human in vitro system that may have relevance to the inflammation that characterizes chronic obstructive pulmonary disease (COPD). To this end, we evaluated the role of IP-receptors in regulating the release from human airway epithelial cells (both primary and the BEAS-2B cell line) of two CXC chemokines: monokine induced by interferon-γ (CXCL9) and interferon-γ-inducible protein of 10 kDa (CXCL10). These CXC chemokines are potent chemoattractants for CD8+ (Tc1) T lymphocytes (Farber, 1997), and they are elevated in the sputum and airways of subjects with COPD (Saetta et al., 2002; Hardaker et al., 2004; Donnelly and Barnes, 2006). Indeed, the abnormal secretion of such CXC chemokines from airway epithelia may account for the high number of pulmonary CD8+ T cells in subjects with COPD compared with normal healthy individuals (O'Shaughnessy et al., 1997; Saetta et al., 1999). Given that CD8+ T cells elaborate perforins and granzyme B (Garcia-Sanz et al., 1988; Chrysofakis et al., 2004), their inappropriate activation may result in secretion of CXCL9 and CXCL10 and promote emphysematous changes in the lung by inducing apoptosis of alveolar epithelial cells (Donnelly and Barnes, 2006).

Chemical structure of RO1138452 (Clark et al., 2004; Bley et al., 2006).
The second aim of this study was to characterize in detail the antagonist properties of RO1138452 and to evaluate the utility of this novel ligand in human IP-receptor classification. For this purpose, BEAS-2B cells stably harboring a cAMP response element (CRE) reporter construct were used because they provide a robust method of interrogating agonist-antagonist interactions.
Materials and Methods
Cell Culture
BEAS-2B cells were cultured for 3 days under a 5% CO2, air atmosphere at 37°C in 6- or 24-well plastic plates containing keratinocyte serum-free medium (KSFM) supplemented with 5 ng/ml epidermal growth factor, 50 μg/ml bovine pituitary extract, 100 mg/ml penicillin, and 100 U/ml streptomycin. The cells were cultured for a further 3 days in fresh KSFM with supplements and then growth-arrested for 24 h in supplement-free KSFM. At this time, cultures were tightly confluent, and they were processed for biochemical and functional measurements as described below.
Human primary airway epithelial cells (HAECs) were obtained by proteinase digestion of nontransplanted normal human lung (International Institute for the Advancement of Medicine, Edison, NJ), as described previously (Churchill et al., 1989). Cells were seeded in 96-well plates (Costar, Cambridge, MA) containing serum-free, bronchial epithelial cell growth medium (BioWhittaker, Walkersville, MD). Cells were cultured under a 5% CO2, air atmosphere at 37°C until confluent (after ∼14 days of culture), growth arrested for 18 h in serum-free, bronchial epithelial cell basal medium, which is devoid of all supplements, and then processed for biochemical and functional measurements as described below. Ethics approval for the use of human tissues has been granted by the Conjoint Health Research Ethics Board of the University of Calgary.
Measurement of CXCL9 and CXCL10
Growth-arrested epithelial cells were treated with IP-receptor agonists/antagonists as indicated in the text and figure legends. IFNγ was then added, and the cells incubated at 37°C under a 5% CO2 atmosphere for 6 to 48 h. The amount of CXCL9 and CXCL10 released into the culture supernatant was quantified by sandwich ELISA (Human DuoSet Development System; R&D Systems, Minneapolis, MN) according to the manufacturer's instructions.
Infection of Cells with Ad5.CMV.PKIα
In some experiments, subconfluent (∼70%), BEAS-2B cells were infected (MOI = 20) with an E1-/E3-replication-deficient adenovirus vector (Ad5.CMV.PKIα) containing a 251-base pair DNA fragment encoding the complete amino acid sequence of the α-isoform of cAMP-dependent protein kinase (PKA) inhibitor (PKI) downstream of the constitutively active CMV immediate early promoter (Meja et al., 2004). After 48 h, cells were processed for the assessment of CXC chemokine release and activation of a CRE reporter construct as described below. To control for biological effects of the virus per se, the vector Ad5.CMV.Null, expressing no transgene, was used in parallel. Using this experimental protocol, we have reported previously that >90% of BEAS-2B cells are infected with Ad5.CMV.PKIα, resulting in the expression of a completely functional transgene with no adverse effect on cell viability (Meja et al., 2004).
Generation of a CRE-Dependent Luciferase Reporter Construct
The plasmid pADneo2-C6-BGL contains six CREs in tandem upstream of a minimal β-globin promoter driving a luciferase gene. BEAS-2B cells at ∼50% confluence in T-75 flasks were transfected with 8 μg of plasmid DNA using Tfx50 (Promega, Madison, WI). After overnight incubation, cells were passaged and cultured in T-162 flasks in the presence of 75 μg/ml G-418 (Geneticin; Invitrogen, Carlsbad, CA). Medium was changed every 2 to 3 days until foci of stable transfectants were noted (after 2 to 3 weeks of culture). These foci were then harvested to create a heterogeneous population of cells in which the sites of plasmid integration were randomized.
Measurement of Luciferase
CRE reporter cells were growth-arrested and treated with IP-receptor agonists/antagonists or salbutamol as indicated in the text and figure legends. Cells were incubated at 37°C under a 5% CO2 atmosphere for 1 to 9 h as indicated, and then they were harvested in reporter lysis buffer; luciferase was then measured according to the manufacturer's instructions. Data are expressed as -fold induction of luciferase relative to unstimulated cells. In some experiments, the IP-receptor agonist iloprost was used. Given that this compound has nanomolar affinity for several Gs-coupled prostanoid receptors (Abramovitz et al., 2000; Wise and Jones, 2000), the culture medium was supplemented with 1 μM BWA 868C, 100 nM L-798,106, and 500 nM L-161,982 to block the DP1-, EP3-, and EP4-receptor subtypes, respectively. Although BEAS-2B cells also express the EP1-receptor subtype, selective agonists neither activate nor inhibit CRE-dependent transcription (unpublished observations). Accordingly, a selective EP1-receptor antagonist was not used in these experiments.
RO1138452 Washout Experiments
BEAS-2B cells were incubated for 30 min at 37°C in supplement-free KSFM in the absence and presence of 100 nM RO1138452. Cells were washed with supplement-free KSFM, incubated in the same medium for defined periods (see Fig. 8), and exposed to 1 μM taprostene. Four hours later, cells were harvested in reporter lysis buffer, and luciferase activity was measured.
Assessment of Cell Viability
The viability HAECs and BEAS-2B cells was determined colorimetrically by measuring the reduction of the tetrazolium salt 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide to formazan, by mitochondrial dehydrogenases.
Curve Fitting
Monophasic agonist concentration-effect (E/[A]) curves were fitted by least-squares, nonlinear iterative regression to the following form of the Hill equation (Prism 4; GraphPad Software Inc., San Diego, CA):  where E is the effect, Emin and Emax are the lower and upper asymptote (i.e., the basal response and maximal agonist-induced response, respectively), p[A] is the log molar concentration of agonist, p[A]50 is a location parameter equal to the log molar concentration of agonist producing Emax/2, and n is the gradient of the E/[A] curve at the p[A]50 level.
where E is the effect, Emin and Emax are the lower and upper asymptote (i.e., the basal response and maximal agonist-induced response, respectively), p[A] is the log molar concentration of agonist, p[A]50 is a location parameter equal to the log molar concentration of agonist producing Emax/2, and n is the gradient of the E/[A] curve at the p[A]50 level.
Characterization of the IP-Receptor Antagonist RO1138452
The nature of the antagonism produced by RO1138452 at IP-receptors was evaluated by least-squares, nonlinear regression. Data derived from the CRE-reporter studies were analyzed using a modification of the Hill and Gaddum/Schild equations derived by Waud et al. (1978). Each family of E/[A] curves (i.e., the control E/[A] curve and E/[A] curves constructed in the presence of increasing concentrations of antagonist) were fitted simultaneously to eq. 2. Thus,  where [A] and [B] are the molar concentration of agonist and antagonist, respectively, S is the Schild slope factor, and pA2 is the affinity of the antagonist when S = 1, which is equivalent to the pKB. To determine whether S deviated significantly from unity, the entire family of E/[A] curves that made up an individual experiment was fitted globally to eq. 2 under two conditions: one condition where S was constrained to a constant equal to 1, and the other condition where it was a shared value for all data sets. The F-test was applied to determine statistically which equation gave the best fit, and this equation was then used for the analysis.
where [A] and [B] are the molar concentration of agonist and antagonist, respectively, S is the Schild slope factor, and pA2 is the affinity of the antagonist when S = 1, which is equivalent to the pKB. To determine whether S deviated significantly from unity, the entire family of E/[A] curves that made up an individual experiment was fitted globally to eq. 2 under two conditions: one condition where S was constrained to a constant equal to 1, and the other condition where it was a shared value for all data sets. The F-test was applied to determine statistically which equation gave the best fit, and this equation was then used for the analysis.
Because complete agonist E/[A] curves could not always be constructed, especially at high antagonist concentrations, the antagonism imposed by RO1138452 of CRE-dependent transcription was also determined using an alternative experimental approach outlined by Lazareno and Birdsall (1993). This was also the preferred method in the chemokine release studies as a detailed Schild analysis was not practicable. Thus, taprostene was used at a fixed concentration of 1 μM (∼p[A]95; ≅10 × p[A]50) in the absence and presence of increasing concentrations of RO1138452. For this type of analysis, knowledge of the precise location of the taprostene E/[A] curve is also required; accordingly, this was determined in parallel on the same batch of cells. The resulting pair of antagonist and taprostene E/[A] curves was then fitted simultaneously to eq. 2. As before, the F-test was applied to determine whether S deviated significantly from unity.
Determination of the Equilibrium Dissociation Constant of Taprostene
The affinity of taprostene (KA) was estimated by operational model fitting (Black and Leff, 1983) using two experimental approaches: following “irreversible” inactivation of a fraction of the total functional receptor population ([Ro]) (Furchgott, 1966) and by comparison of taprostene E/[A] curves with those of the full reference agonist iloprost (Barlow et al., 1967).
Receptor Inactivation.E/[A] curves were generated to taprostene in the absence and presence of concentrations of RO1138452 that depressed the upper asymptote, Emax, of the control curve. For this type of experiment only a single E/[A] curve can be determined per batch of cells. Accordingly, the data (i.e., all E/[A] curves constructed in the absence and presence of either a single concentration of antagonist or every concentration of antagonist), were fitted simultaneously to eq. 3 (Leff et al., 1990c), which describes a theoretical relation between pharmacological effect and agonist concentration (Black and Leff, 1983). Thus,  where Em is the theoretical maximal response of the tissue (i.e., maximal effect produced by a full agonist), and τ is the operational efficacy of the agonist, which is the ratio of [Ro] to the concentration of agonist-receptor complexes ([AR]) required to produce half-maximal effect ([KE]) (Leff et al., 1990c). This analysis yielded a single estimate of Em, n, and KA as well as an efficacy value for agonist before (τ′) and after (τ) receptor inactivation. The percentage of functionally active receptors (q) remaining after treatment of cells with RO1138452 is given by (τ/τ′) × 100.
where Em is the theoretical maximal response of the tissue (i.e., maximal effect produced by a full agonist), and τ is the operational efficacy of the agonist, which is the ratio of [Ro] to the concentration of agonist-receptor complexes ([AR]) required to produce half-maximal effect ([KE]) (Leff et al., 1990c). This analysis yielded a single estimate of Em, n, and KA as well as an efficacy value for agonist before (τ′) and after (τ) receptor inactivation. The percentage of functionally active receptors (q) remaining after treatment of cells with RO1138452 is given by (τ/τ′) × 100.
Comparative Method.E/[A] curves were constructed to taprostene, which is a partial agonist in BEAS-2B cells, and the full agonist iloprost. The experiment was conducted nine times and the reference full agonist and partial agonist E/[A] curves were fitted simultaneously to eqs. 1 and 3, respectively.
Determination of Receptor Reserve
Receptor occupancy-effect curves were constructed to taprostene using the KA determined by receptor “inactivation” and the comparative method. At each concentration of agonist and, therefore, at each level of response, fractional IP-receptor occupancy [i.e., the ratio of agonist occupied IP-receptors to the total number of available receptors (RA/Rt)] in untreated, “control” cells was determined applying eq. 4 (Furchgott, 1966), where 
As each E/[A] curve was constructed to taprostene in half-log increments of concentration, the point at which Emax is achieved cannot be determined with accuracy. Thus, an estimate of this value was obtained by determining the fraction of agonist-occupied IP-receptors that elicited 95% of the maximal response.
Drugs and Analytical Reagents
RO1138452 (a.k.a. CAY 10441), BWA 868C, iloprost, and taprostene were from Cayman Chemical (Ann Arbor, MI). L-161,982 and L-798,106 were donated by Merck Frosst (Montreal, QC, Canada). All other chemicals and reagents were from Sigma-Aldrich (Oakville, ON, Canada). Prostanoid agonists and antagonists were dissolved in dimethyl sulfoxide, and they were diluted to the desired working concentration in the appropriate culture medium. None of the compounds or their vehicles used in the experiments described herein significantly affected cell viability using the reduction of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide to formazan as an indicator.
General Statistics
Data points, and values in the text and figure legends, represent the mean ± S.E.M. of n independent determinations. Where appropriate, data were analyzed statistically using Student's paired t test or by one-way analysis of variance/Newman-Keuls multiple comparison test. The null hypothesis was rejected when p < 0.05.
Results
Chemokine Release
IFNγ-Induced CXCL9 and CXCL10 Release from BEAS-2B Cells. The amount of CXCL9 and CXCL10 released spontaneously from BEAS-2B cells after they had been cultured for 24 h in KSFM (without supplements) was below the detection limit of the assays. In contrast, cells exposed to IFNγ (0.1 ng/ml–3 μg/ml) released CXCL9 and CXCL10 into the culture supernatant in a concentration-dependent manner, with p[A]50 values (grams per milliliter) of -7.32 ± 0.2 and -7.53 ± 0.14, respectively (p > 0.05; Fig. 2a). At a near maximally effective concentration of IFNγ (3 μg/ml), CXCL9 and CXCL10 were present in the culture medium at concentrations of 1.21 ± 0.18 and 1.58 ± 0.33 ng/ml, respectively (p > 0.05; Fig. 2a). Unless stated otherwise, IFNγ was used in all further experiments with BEAS-2B cells at a concentration of 100 ng/ml, which equated to the p[A]60 for the release of both chemokines.
Kinetic studies established that CXCL9 and CXCL10 were released into the culture medium after a lag of approximately 4 h. Thereafter, the amount of both chemokines increased steadily, with t1/2 values of 29.9 ± 4.1 h (CXCL9) and 18.2 ± 0.8 h (CXCL10; p < 0.05). The amount of CXCL10 peaked at approximately 30 h after exposure to IFNγ, and no further change was detected up to 48 h (Fig. 2b). In contrast, the time course of CXCL9 release was more protracted and the maximum response could not accurately be defined (Fig. 2b).
Effect of Taprostene on CXCL9 and CXCL10 Release from BEAS-2B Cells. Taprostene (1 nM–10 μM) inhibited, in a concentration-dependent manner, the release of CXCL9 from IFNγ-treated BEAS-2B cells, with a p[A]50 (molar) and Emax of -7.31 ± 0.07 and 32.4 ± 1.1%, respectively (n = 18). Statistically indistinguishable data were obtained for taprostene on the output of CXCL10 (p[A]50 =-7.40 ± 0.05; Emax = 30.3 ± 0.9%; n = 21; p > 0.05). These inhibitory effects were abolished in cells infected with the PKIα expression vector, Ad5.CMV.PKIα (MOI = 20), but not the empty virus, Ad5.CMV.Null (Fig. 3, a and c).
Effect of 8-Br-cAMP on CXCL9 and CXCL10 Release from BEAS-2B Cells. Pretreatment (30 min) of BEAS-2B cells with 30 μM 8-Br-cAMP attenuated the ability of 100 ng/ml IFNγ to release CXCL9 and CXCL10 by 31.1 and 55.4%, respectively (Fig. 3, b and d). This inhibitory effect was abolished in cells infected with Ad5.CMV.PKIα (MOI = 20) but not Ad5.CMV.Null (Fig. 3, b and d). Neither virus vector influenced the release of CXCL9 and CXCL10 evoked by IFNγ (Fig. 3, b and d).

Concentration-dependence and kinetics of IFNγ-induced CXCL9 and CXCL10 release from BEAS-2B cells. a, growth-arrested cells were treated with IFNγ (0.1 ng/ml–3 μg/ml) for 24 h, and the amount of CXCL9 and CXCL10 released into the culture supernatant was determined by ELISA. b, growth-arrested cells were treated with a fixed concentration of IFNγ (100 ng/ml), and the concentration of CXCL9 and CXCL10 in the culture supernatant was determined at defined times over a period of 48 h. Data points in a and b represent the mean ± S.E.M. of eight and four independent determinations, respectively.

Effect of PKIα on the inhibition by taprostene and 8-Br-cAMP of CXCL9 and CXCL10 release from BEAS-2B cells. Naive and Ad5-infected BEAS-2B cells (MOI = 20; 48 h) were pretreated (30 min) with taprostene (1 nM–10 μM; a and c) or 8-Br-cAMP (30 μM; b and d). IFNγ (100 ng/ml) was then added, and after 24 h the amount of CXCL9 (a and b) and CXCL10 (c and d) released into the culture supernatant was quantified by a sandwich ELISA. Data in each panel represent the mean ± S.E.M. of three independent determinations. *, p < 0.05, significant inhibition of chemokine release.
Effect of RO1138452 on Taprostene-Induced Inhibition of CXCL9 and CXCL10 Release from BEAS-2B Cells. RO1138452 had no effect on IFNγ-induced chemokine release from BEAS-2B cells at any concentration examined. In contrast, RO1138452 (10 pM–10 μM) added to cells concurrently with a fixed concentration of taprostene (1 μM) prevented, in a concentration-dependent manner, the inhibition of CXCL9 and CXCL10 release, with p[A]50 (molar) values of -8.73 ± 0.11 and -8.47 ± 0.16 (p > 0.05), respectively (Fig. 4, a and b). Enumeration of the Schild slope factor, S, by simultaneously fitting to eq. 2 each RO1138452 and taprostene E/[A] curve indicated that this parameter deviated significantly (p < 0.05) from unity for the antagonism of both CXCL9 (S = 0.745 ± 0.05; n = 9) and CXCL10 (S = 0.674 ± 0.08; n = 9). Thus, RO1138452 behaved in a manner that was inconsistent with surmountable competitive antagonism (Neubig et al., 2003).

Waud/Birdsall/Lazareno analysis of the antagonism by RO1138452 of taprostene-induced inhibition of CXCL9 and CXCL10 release from BEAS-2B cells. Growth-arrested cells were pretreated (30 min) concurrently with RO1138452 (10 pM–1 μM) in the presence of 1 μM taprostene, or with taprostene (1 nM–10 μM) alone. IFNγ (100 ng/ml) was then added, and after 24 h the amount of CXCL9 (a) and CXCL10 (b) released into the culture supernatant was quantified by a sandwich ELISA. Each pair of E/[A] curves was then fitted simultaneously to eq. 2 from which the Schild slope factor (S) was derived. Data in each panel represent the mean ± S.E.M. of nine independent determinations.
IFNγ-Induced CXCL9 and CXCL10 Release from HAECs: Effect of Taprostene and Antagonism by RO1138452. The amount of CXCL9 and CXCL10 released spontaneously from HAECs after they had been cultured for 18 h in serum-free, bronchial epithelial cell basal medium was routinely low or below the detection limit of the ELISAs (Fig. 5, a and b). In contrast, exposure of cells to IFNγ (0.1–300 ng/ml) resulted in a robust, concentration-dependent release of CXCL9 and CXCL10 into the culture supernatant [p[A]50 values (grams per milliliter) =-8.15 ± 0.09 and -8.25 ± 0.12, respectively; p > 0.05; Fig. 5, a and b]. At the highest concentration of IFNγ (300 ng/ml) studied, which was equivalent to the p[A]95 predicted by eq. 1, CXCL9 and CXCL10 were present in the culture medium in amounts (12.1 ± 1.7 and 12.0 ± 0.54 ng/ml, respectively; p > 0.05; Fig. 5, a and b) that were ∼10-fold higher than that produced by BEAS-2B cells under similar cell culture conditions (see Fig. 2a). As shown in Fig. 5, a and b, concurrent treatment of cells with IFNγ and taprostene (1 μM) significantly depressed the upper asymptote, Emax, of the mean E/[A] curves that described the release of both chemokines without significantly affecting potency [p[A]50 (grams per milliliter): CXCL9 =-8.19 ± 0.09; CXCL10 =-8.32 ± 0.21; p > 0.05]. Exposure of HAECs to 30 μM 8-Br-cAMP also attenuated the ability of IFNγ to release CXCL9 and CXCL10 by 58.9 and 50.1%, respectively (Fig. 5, c and d).
RO1138452 had no effect on IFNγ-induced chemokine release from HAECs at any concentration examined. However, when added to cells concurrently with a fixed concentration of taprostene (1 μM), RO1138452 (100 pM–1 μM) reversed, in a concentration-dependent manner, the inhibition of CXCL9 and CXCL10 release, with mean p[A]50 (molar) values of -7.88 and -8.27, respectively (Fig. 5, e and f). Consistent with the data shown in Fig. 4, these E/[A] curves were very shallow, with Hill coefficients considerably less than unity (nH: CXCL9 = 0.313; CXCL10 = 0.425).
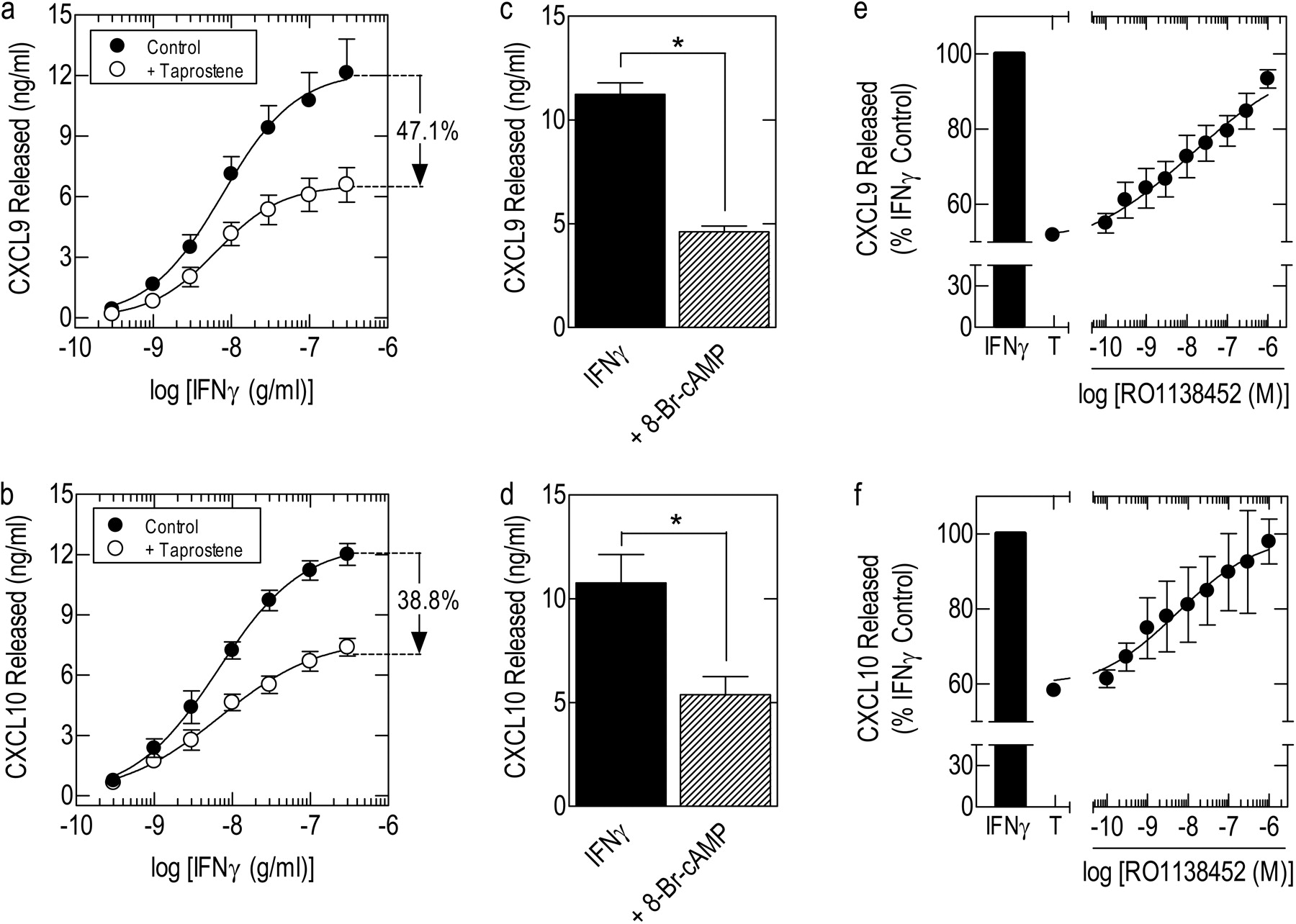
Effect of taprostene and 8-Br-cAMP on chemokine release from HAECs and antagonism by RO1138452. a and b, growth-arrested cells were pretreated (30 min) with 1 μM taprostene or vehicle, and then they were exposed to IFNγ (0.1–300 ng/ml). c and d, cells were pretreated (30 min) with 30 μM 8-Br-cAMP, and then they were exposed to a fixed concentration of IFNγ (100 ng/ml). e and f, cells were pretreated (30 min) with taprostene (T; 1 μM) in the absence and presence of RO1138452 (10 pM–1 μM), and then they were exposed to 100 ng/ml IFNγ. In each experiment, the amount of CXCL9 (a, c, and e) and CXCL10 (b, d, and f) released into the culture supernatant at 24 h was determined by ELISA. Data represent the mean ± S.E.M. of nine independent determinations using cells harvested from four donors (a and b) and four independent determinations using cells harvested from four donors (c–f). *, p < 0.05, significant inhibition of chemokine release.
CRE-Dependent Transcription
Effect of Taprostene on CRE-Dependent Transcription. Treatment of BEAS-2B cells stably harboring a CRE reporter with taprostene (100 nM, 1 μM, and 10 μM) induced the luciferase gene in a time-dependent manner (Fig. 6a). The kinetic of this effect was independent of agonist concentration [t1/2(on) ∼1.7 h], reached a maximum at the 4 to 5-h time point, and thereafter it decayed toward baseline levels [t1/2(off) ∼ 3.5 h]. At the 5-h time point, the induction by taprostene of the luciferase gene was concentration-related, with a p[A]50 (molar) and maximal -fold induction, Emax, over untreated cells of -7.08 ± 0.03 and 3.60 ± 0.10, respectively (n = 48). Infection of cells with Ad5.CMV.PKIα (MOI = 20) abolished taprostene-induced CRE-dependent transcription, whereas Ad5.CMV.Null was inactive (Fig. 6b).

Kinetics and concentration dependence of taprostene-induced activation of a CRE-reporter construct stably expressed in BEAS-2B cells. a, growth-arrested cells were exposed to taprostene (0.1, 1, and 10 μM), and the expression of luciferase was measured every 60 min over a period of 9 h. b, cells were infected with Ad5.CMV.PKIα, Ad5.CMV.Null, or left untreated (naive), and then they were exposed to taprostene (1 nM–10 μM). Luciferase was measured at 5 h. Data in a and b represent the mean ± S.E.M. of four and three independent determinations, respectively.
Characterization of the IP-Receptor Antagonist RO1138452. Pretreatment (30 min) of BEAS-2B cells with 100 nM RO1138452 had no effect on the basal expression of luciferase, but it dramatically depressed (by 71.4% at 10 μM) the upper asymptote, Emax, of the mean taprostene E/[A] curve (Fig. 7a). This effect was agonist independent; indeed, a similar reduction in Emax (70% at 1 μM) was produced when iloprost was substituted for taprostene (Fig. 7b). The ability of RO1138452 to reduce the taprostene Emax was concentration-related, and it was accompanied by a graded dextral shift of the E/[A] curves and an associated (up to ∼1.3 log10 units at 300 nM) reduction in agonist potency (Fig. 7c; Table 1).
Effect of the IP-receptor antagonist RO1138452 on the potency and ability of taprostene to induce a CRE-driven luciferase gene stably expressed in BEAS-2B epithelial cells Cells were incubated with RO1138452 or vehicle for 30 min at the concentrations indicated. Taprostene E/[A] curves were then constructed in the presence of antagonist and fitted to eq. 1 from which estimates of agonist potency, p[A]50, and -fold induction (Emax) of the luciferase gene were interpolated. Data represent the mean ± S.E.M. of five independent determinations.
RO1138452 (100 nM) also dramatically reduced Emax when added to cells concurrently with taprostene (30.5%) or iloprost (43.3%) and luciferase measured at 5 h (Fig. 7, a and b). However, in these experiments the magnitude of Emax depression was less than when cells were pretreated with the antagonist before being exposed to taprostene or iloprost (Fig. 7, a and b). Increasing the time cells were exposed to RO1138452 in combination with taprostene or iloprost in an attempt to achieve equilibrium conditions before reporter activity was measured was impracticable, because the “signal-to-noise” ratio was too high at time points beyond 7 h due to a rapid decline of the luciferase signal (Fig. 6a).
Figure 7d shows the effect on CRE-dependent transcription of RO1138452 (10 pM–10 μM) added to cells concurrently with a fixed concentration of taprostene (1 μM). Using this alternative experimental approach, RO1138452 prevented, in a concentration-dependent manner, the activation of the reporter construct. However, Waud/Lazareno/Birdsall analysis could not be performed, because the RO1138452 E/[A] curves in the presence of taprostene were biphasic, with p[A]50 values for the high- and low-affinity components of -8.95 ± 0.46 (57.6 ± 7.1%) and -6.81 ± 0.15, respectively. Thus, using CRE-dependent transcription as a functional output, RO1138452 also behaved in a manner that was inconsistent with surmountable competitive antagonism (Neubig et al., 2003).

Effect of RO1138452 on taprostene- and iloprost-induced activation of the CRE reporter construct stably expressed in BEAS-2B cells. a and b, growth-arrested cells were exposed to taprostene (1 nM–10 μM) or iloprost (100 pM–1 μM) concurrently (C) with RO1138452 (100 nM) or after pretreatment (P; 30 min) with the antagonist. c, cells were pretreated (30 min) with RO1138542 (10, 30, and 300 nM), and then they were exposed to taprostene (1 nM–10 μM). d, growth-arrested cells were exposed (30 min) concurrently to RO1138452 (10 pM–1 μM) in the presence of taprostene (1 μM) or to taprostene (1 nM–10 μM) alone. In all experiments, cells were processed at 5 h, and luciferase activity was determined. Each data set of E/[A] curves in c was fitted globally to eq. 3, to give model parameter estimates (see Table 2). In d, Waud/Birdsall/Lazareno analysis could not be performed because the RO1138452 E/[A] curves in the presence of taprostene were biphasic. Data in a, b, c, and d represent the mean ± S.E.M. of four, six, five, and six paired experiments, respectively.
An additional set of washout experiments was conducted to gauge the reversibility of RO1138452. BEAS-2B cells were pretreated (30 min) with 100 nM RO1138452 or vehicle, washed with antagonist-free medium (3 × 2.5 ml), and then incubated in fresh medium for 0 to 20 h as indicated. Taprostene (1 μM) was added, and luciferase was measured at 5 h. As shown in Fig. 8, taprostene activated the reporter ∼3-fold at all time points when measurements were made, and this response was significantly antagonized by RO1138452 before “washout” (i.e., at time 0) and at all time points thereafter. Indeed, at 20 h, taprostene-induced CRE-dependent transcription was suppressed by 55.5 ± 17.8%, and this was not significantly different (p > 0.05) from the antagonism produced at time 0 (84.2 ± 9.4%).
Effect of RO1138452 on β2-Adrenoceptor-Mediated, CRE-Dependent Transcription. Treatment of BEAS-2B CRE reporter cells with the selective β2-adrenoceptor agonist salbutamol (1 μM) increased transcription 8.16 ± 0.72-fold (n = 3). RO1138452 (100 nM), added concurrently with salbutamol, failed to significantly antagonize this response (7.83 ± 0.84-fold; n = 3; p > 0.05) under conditions where taprostene-induced transcription was markedly attenuated (Fig. 7a).
Estimation of the KA of Taprostene for the Human IP-Receptor: Inactivation Method. The insurmountable antagonism produced by RO1138452 in BEAS-2B cells at 5 h was exploited to estimate the affinity of taprostene for the IP-receptor subtype using operational curve fitting (Tables 2 and 3). Each data set that makes up Fig. 7c (i.e., the control E/[A] curve and the associated E/[A] curves obtained at each concentration of antagonist) was fitted globally to eq. 3, from which a mean pKA of -5.89 (KA = 1.3 μM) was determined (Table 3). Analysis of the control E/[A] curves relative to those generated at each individual concentration of antagonist showed that the affinity of taprostene estimated in this way was essentially invariant irrespective of the concentration of RO1138452 used to depress Emax and that it differed by only 0.3 log10 units from the pKA (-6.19) estimated from the simultaneous analysis of E/[A] curves at all concentrations of antagonist (Table 2). Operational curve fitting implied that taprostene had reasonable efficacy in this system (mean τ' = 14.3; Leff et al., 1990c), but, nevertheless, it was a partial agonist relative to the estimated Em, with an intrinsic activity (α), calculated as the ratio of (Emax - 1)/(Em - 1), of 0.84 (Tables 1 and 2).
Estimates of pKA, Emax, n,pτ′,pτ, and q for the effect of RO1138452 on taprostene-induced activation of a CRE-driven luciferase gene stably expressed in BEAS-2B airway epithelial cells Cells were incubated with RO1138452 or vehicle for 30 min at the concentrations indicated in the table. Taprostene E/[A] curves were then constructed in the continued presence of antagonist, and the data were fitted to eq. 3 as described under Materials and Methods. Data represent the mean ± S.E.M. of five independent determinations.
Estimates of pKA, Em, n, and pτ′ determined by the inactivation and comparative methods on taprostene-induced activation of a CRE-driven luciferase gene stably expressed in BEAS-2B airway epithelial cells

RO1138452 washout experiments. BEAS-2B cells in 24-well plates were incubated for 30 min in medium containing 100 nM RO1138452 or vehicle, washed (3 × 2.5 ml) with antagonist-free medium, and incubated in 2.5 ml of the same medium for 0 to 20 h. Taprostene (1 μM) was then added, and 5 h later cells were harvested in reporter lysis buffer, and luciferase activity was measured. Data represent the mean ± S.E.M. of four independent paired determinations.
Estimation of KA of Taprostene for the Human IP-Receptor: Comparative Method. The IP-receptor agonist iloprost (0.1 nM–1 μM) activated the CRE reporter in a concentration-dependent manner. As shown in Fig. 9, iloprost was ∼15-fold more potent (p[A]50 =-8.25 ± 0.13; p < 0.05) than taprostene, and it was more effective. Thus, taprostene was a partial agonist in this system, with an intrinsic activity [i.e., Emax - 1(taprostene)/Emax - 1(iloprost))] of 0.82, which was similar to that predicted by the inactivation method (α = 0.84). Simultaneously fitting each pair of taprostene and iloprost E/[A] curves that make up Fig. 9 to eqs. 1 and 3, respectively, yielded for taprostene pKA,pτ', n, and Em estimates that were very similar to the same parameters determined by receptor inactivation (Table 3).
Relationship between IP-Receptor Occupancy and CRE Activation. Using the KA of taprostene estimated by the inactivation method (1.3 μM), IP-receptor occupancy expressed as a function of response was described by a curvilinear relationship that deviated significantly from the line of identity (where response is directly proportional to occupancy; Fig. 10). Thus, an IP-receptor “reserve” for taprostene was present in BEAS-2B cells at submaximal responses (Fig. 10; Table 4). However, the “spare receptors” in this system declined incrementally with concentration and, therefore, magnitude of response, and they were almost depleted at the Emax (1.6-fold excess at the p[A]95; Fig. 10; Table 4). The IP-receptor occupancy/response relationship was also curvilinear using the KA estimated by the comparative method (851 nM), and it was essentially indistinguishable from the affinity of taprostene estimated by receptor inactivation (Fig. 10). Thus, both methods indicated an IP-receptor reserve for taprostene in BEAS-2B cells at low levels of response that became limiting toward the maximal asymptote (Fig. 10; Table 4).
IP-receptor occupancy/response relationship for taprostene-induced activation of CRE-dependent transcription
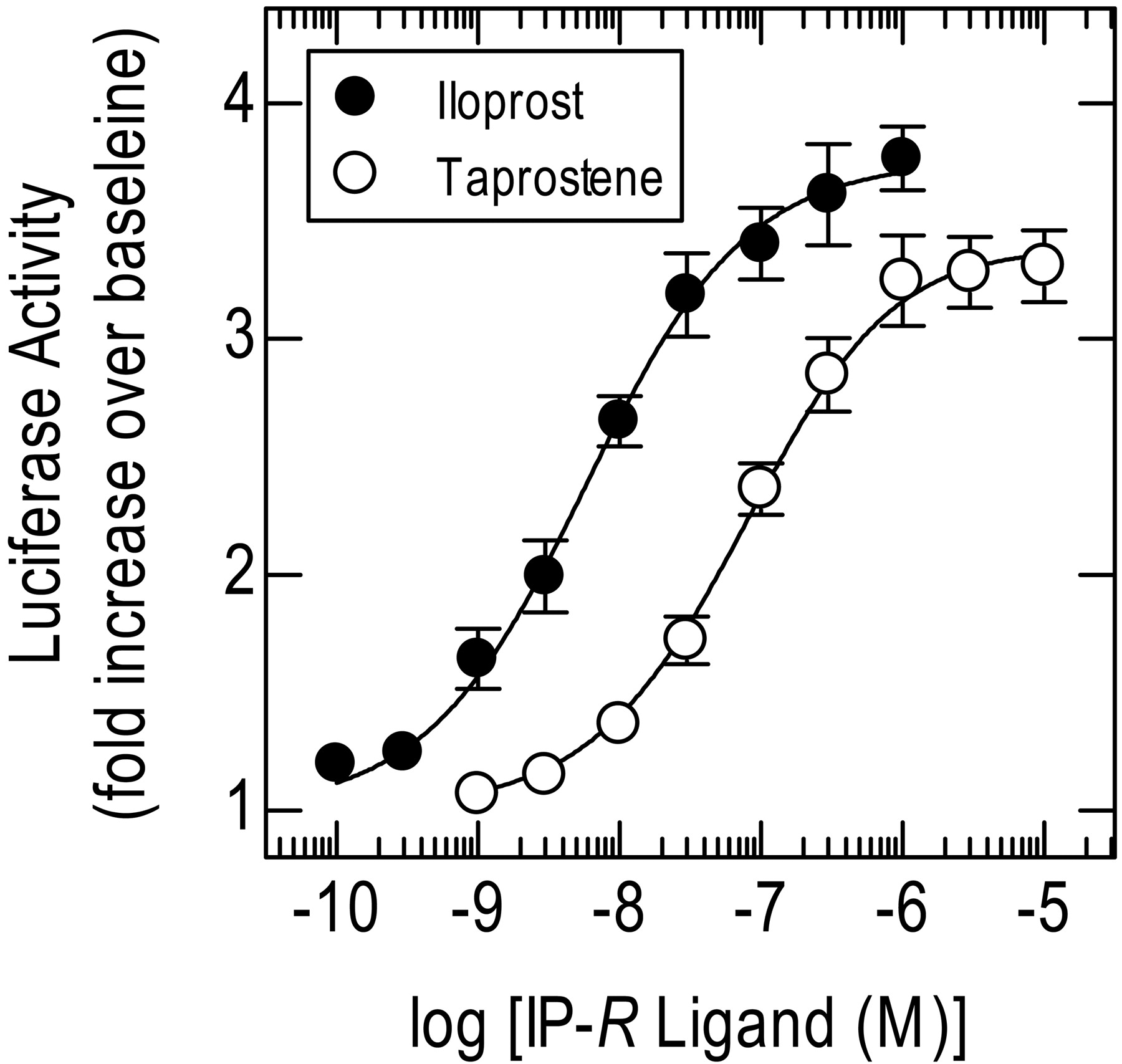
Analysis of taprostene E/[A] curve data by comparison with the reference full agonist iloprost. E/[A] curves were constructed in parallel to taprostene (1 nM–10 μM) and iloprost (100 pM–1 μM) for activation of CRE-dependent transcription. Each resulting pair of taprostene and iloprost E/[A] curves was then fitted simultaneously to eqs. 1 and 3, respectively. See Table 3 for model parameter estimates. Data represent the mean ± S.E.M. of nine independent paired determinations. All experiments with iloprost were performed in medium supplemented with 1 μM BWA 868C, 100 nM L-798,106, and 500 nM L-161,982 to block the DP1-, EP3-, and EP4-receptor subtypes, respectively.
Discussion
Many IP-receptor agonists have affinity at multiple prostanoid receptors (see Introduction). Accordingly, responses they elicit cannot be considered diagnostic of IP-receptor agonism. This lack of selectivity greatly hinders the design of experiments and interpretation of data in which the characterization of IP-receptor antagonists in a particular tissue is a primary objective. To circumvent these problems, we used in the present study the stable PGI2 analog taprostene (Schneider et al., 1993). This ligand is selective for the IP-subtype at concentrations up to 10 μM (Chan and Jones, 2004; Supplemental Fig. 1); therefore, it can be used in tissues expressing multiple prostanoid receptors.
Anti-Inflammatory Potential of IP-Receptor Agonists. Airway epithelial cells can elaborate a plethora of proinflammatory mediators (Saetta et al., 2002), and for this reason they are implicated in the pathogenesis of COPD. Herein, we report that taprostene suppressed the output, from BEAS-2B cells, of two CXC chemokines that are implicated in pulmonary CD8+ T-lymphocyte recruitment. Furthermore, RO1138452 antagonized these effects of taprostene at concentrations that selectively target the IP-receptor (Bley et al., 2006, Jones et al., 2006). RO1138452 also blocked the effect of taprostene on HAECs, indicating that data obtained from BEAS-2B cells on IP-receptor function may reliably be extrapolated to primary airway epithelia. Further studies established that PKIα abolished the effects of taprostene and 8-Br-cAMP on chemokine release and CRE-dependent transcription (see below), indicating that human airway epithelial cells express inhibitory IP-receptors coupled to the activation of the cAMP/PKA cascade.
RO1138452 Is a Pseudo-Irreversible Orthosteric Antagonist. On BEAS-2B cells, RO1138452 did not antagonize the actions of taprostene in a competitive manner (Schild slopes <1). The interaction of RO1138452 with the IP-receptor on HAECs was also complex as evinced by the very shallow Hill slopes associated with the reversal of CXCL9 and CXCL10 release. Therefore, the basis for this antagonism was investigated further using BEAS-2B cells stably transfected with a CRE-reporter construct. This experimental system was selected as an output over chemokine release because it produces highly robust and reproducible data; thus, it is ideally suited for the interrogation of agonist/antagonist interactions. On CRE-dependent transcription, RO1138452 also behaved insurmountably. Thus, taprostene and iloprost E/[A] curves were displaced dextrally, and Emax was depressed. In contrast, β2-adrenoceptor-mediated transcription was unaffected by RO1138452, confirming that this antagonist selectively interacts with the IP-receptor subtype (Clark et al., 2004; Bley et al., 2006). Further evidence for a deviation from simple competitive behavior was our inability to perform Waud/Birdsall/Lazareno analysis because RO1138452 E/[A] curves in the presence of taprostene (1 μM) were biphasic (see below for discussion of this effect).

Relationship between IP-receptor occupancy and activation of CRE-dependent transcription. The KA values derived by the inactivation and comparative methods were used to calculate the fractional receptor occupancy (RA/Rt; eq. 4) for taprostene (1 nM–10 μM), and then they were plotted against defined increments of response.
Several mechanisms can account for insurmountable antagonism, including covalent receptor inactivation, pseudo-irreversible binding, allosterism, and conditions where equilibrium among agonist, antagonist, and receptor are incomplete at the time a given response is measured. RO1138452 does not contain any obvious reactive moieties, implying that the depression of Emax is not due to depletion of functional IP-receptors. A state of hemiequilibrium is also unlikely to account for these results. If such a mechanism was operative, RO1138452 should have displaced taprostene E/[A] curves to the right, similar to the behavior of a competitive antagonist, and also reduced Emax to a new steady-state level reflecting a situation where agonist, antagonist, and receptor have partially re-equilibrated (Kenakin et al., 2006). Reference to Fig. 7c shows that RO1138452 did not produce this pattern of antagonism. Indeed, RO1138452 depressed Emax in a concentration-dependent manner that was associated with a progressive dextral displacement of the taprostene E/[A] curve. Several pieces of evidence also argue against an allosteric interaction (i.e., the binding of RO1138452 to a site within the receptor that is topographically distinct from where agonists interact). At saturating concentrations, an allosteric inhibitor often incompletely suppresses the functional response of interest. In this scenario, a condition is reached where increasing the concentration of antagonist further fails to produce any additional dextral displacement of the agonist E/[A] curve or depression of Emax (Kenakin et al., 2006). Thus, receptor signaling remains partially intact, and this accounts for the residual functional response that is measured. The finding that RO1138452 abolished CRE-dependent transcription (Fig. 7d) clearly excludes this mode of antagonism. An allosteric antagonist can also completely block receptor signaling and so abolish functional responses mediated by that receptor. A prediction of this form of antagonism is that the allosteric ligand, which by definition acts at a site distinct from the agonist binding domain, should produce the same effect on Emax and on the location of the E/[A] curve regardless of whether it is added before, or together with, agonist. The data shown in Fig. 7, a and b, shows that the depression of Emax and degree of dextral displacement of iloprost and taprostene E/[A] curves were significantly greater when cells were pretreated with RO1138452 before agonist exposure than when agonist and antagonist were added concurrently. Thus, RO1138452 did not behave in a manner consistent with an allosteric antagonist that abolishes receptor signaling. On the balance of available evidence, we conclude that RO1138452 probably behaves as an orthosteric insurmountable antagonist (i.e., directly interacts with the primary agonist binding site to preclude access for activating ligands) at the human IP-receptor on human airway epithelial cells. The relatively modest reduction in Emax produced by RO1138452 when added to cells concurrently with agonist (Fig. 7, a and b) compared with cells subjected to an antagonist pretreatment protocol indicates that the insurmountability is only apparent. This type of behavior usually arises with antagonists that dissociate very slowly from their cognate receptor such that agonist, antagonist, and receptor are not in equilibrium at the time the response of interest is measured (Vauquelin et al., 2002). Indeed, washout studies established that RO1138452 was a long-acting antagonist, which might be explained by a tight binding, pseudo-irreversible interaction with the IP-receptor. Moreover, the finding that the degree of antagonism imposed by RO1138452 was not reversed after a 20-h washout period is consistent with this tenet and would also account for the lack of competitive behavior when chemokine release (measured at 24 h) was used as a functional output. Thus, on the BEAS-2B cell IP-receptor, our data strongly suggest that RO1138452 behaved pseudo-irreversibly. Given that equilibrium is not attained under these conditions, the affinity of RO1138452 cannot be determined (Kenakin et al., 2006).
The mechanism for the long-lasting interaction of RO1138452 with the IP-receptor was not investigated in this study. However, the biphasic RO1138452 E/[A] curve that described the antagonism of taprostene-induced transcription (Fig. 7d) has been documented previously for the interaction of certain antagonists with the type 1 angiotensin II receptor in vascular smooth muscle (Vanderheyden et al., 2000). In those studies, it was proposed that this pharmacological profile represented an equilibrium between “tight binding/slowly reversible” and “relatively weak binding/rapidly reversible” states of the antagonist/receptor complex. Indeed, mathematical modeling of such a mechanism was shown subsequently to readily account for the behavior of these antagonists at the type 1 angiotensin II receptor (see Lee et al., 2007, and references therein). Therefore, it is possible that this two-state model could also help explain the interaction of RO1138452 with the IP-receptor.
RO1138452 Is a Competitive Surmountable Antagonist in Other Tissues. The insurmountability of RO1138452 was unexpected given its competitive behavior in other tissues (Bley et al., 2006; Jones et al., 2006). However, such conflicting data are not unprecedented. For example, the 5-hydroxytryptamine2-receptor antagonist methysergide behaves competitively and noncompetitively across different tissues (even from the same species) that are thought to express the same 5-hydroxytryptamine2-receptor (see Vauquelin et al., 2002, and references therein).
Several explanations could account for this anomaly. Differences in sequence between orthologous GPCRs that do not affect the pharmacology of endogenous agonists may, nevertheless, modify the interaction of synthetic ligands. Multiple receptor subtypes expressed variably across tissues could also give rise to insurmountability. However, both of these possibilities are improbable given that RO1138452 is an apparently competitive antagonist in some tissues of human origin (Jones et al., 2006) and IP-receptor heterogeneity has not convincingly been demonstrated. Two other theories are relevant. First, the cellular microenvironment in which a GPCR is expressed can influence the behavior of antagonists (Kenakin, 2003). Such differences are, thus, tissue-dependent, and antagonists will, therefore, have distinct “phenotypical” profiles. Whether tissue-dependent factors can alter the behavior of RO1138452 is unknown, but our data do not allow phenotypical considerations to be excluded. Second, the presence of a substantial IP-receptor reserve in a tissue may mask, initially, the ability of an antagonist to act insurmountably (Vauquelin et al., 2002). In such tissues, which could include many of those studied by Jones et al. (2006), a full agonist will still elicit the maximum response in the presence of RO1138452 because it needs to occupy only a small fraction of the receptor population. Thus, the agonist E/[A] curve is displaced to the right without depression of Emax. Only when the concentration of RO1138452 is increased to a level where IP-receptor number becomes limiting is insurmountable behavior seen. This theoretical scenario contrasts with the results described herein where taprostene was a partial agonist on CRE-dependent transcription and IP-receptor number was limiting. Hence, RO1138452, even at very low concentrations, produced a dextral displacement of the taprostene E/[A] curve and concomitantly depressed Emax. It is noteworthy that Jones et al. (2006) have reported that the Emax of cicaprost (IP-agonist) for the relaxation of rabbit mesenteric artery and guinea pig aorta was slightly depressed (by ∼20%) in the presence of 100 nM and 1 μM RO1138452, respectively. Although these investigators attributed this effect to functional antagonism (i.e., the activation of contractile EP3-receptors by high agonist concentrations), the data could also be explained if RO1138452 bound pseudo-irreversibly to the IP-receptor under conditions where it had depleted the receptor reserve for cicaprost.
Estimation of the KA of Taprostene and Receptor Reserve. An antagonist that behaves pseudo-irreversibly should behave identically to an irreversible competitive antagonist (Kenakin, 1984). Accordingly, RO1138452 was exploited to estimate the affinity of taprostene for the human IP-receptor and to determine the relationship between receptor occupancy and response. Estimating the affinity of an agonist for a GPCR using pharmacological means is theoretically invalid due to the operation of ternary complex mechanisms (Leff et al., 1990a; Colquhoun, 1998). Indeed, theory predicts that the method of receptor inactivation overestimates affinity by a factor proportional to agonist intrinsic efficacy. Therefore, in the present study, the KA of taprostene was also determined by the comparative method, using iloprost as a reference full agonist. By definition, the analysis of partial agonists is considerably less prone to error, and it provides a close approximation of the true KA if its intrinsic efficacy is low relative to the full agonist. As described by Leff and Harper (1989), simultaneous application of these two methods provides an experimental “check” for the operation of ternary complex mechanisms because these independent estimates of affinity should not correspond.
Using this practical test, the KA of taprostene was estimated to be 1.3 and 0.85 μM using the receptor inactivation and comparative methods, respectively. Thus, in this experimental system, there was no indication that the receptor inactivation method introduced the error in affinity predicted by theory. Similar results have been reported in other tissues (Waud, 1969; Leff et al., 1990b), indicating that under certain circumstances, pharmacological methods can provide reliable estimates of affinity despite the operation of ternary complex mechanisms (Leff et al., 1990a). Moreover, the KA of taprostene for the human IP-receptor reported herein is very similar to its pA2 (6.1) determined using the Schild equation for the antagonism of IP-receptor-mediated relaxation of rabbit saphenous vein (Jones and Chan, 2005). However, species differences in IP-receptor pharmacology may be apparent since the affinity of taprostene is 6- to 25-fold higher for the relaxant IP-receptor expressed in pig and guinea pig saphenous veins (pA2 = 7.4 and 6.8, respectively) (Jones and Chan, 2005).
The KA of taprostene estimated by both pharmacological methods was then used to establish the relationship between IP-receptor occupancy and response. Irrespective of whether the KA was set to a value of 1.3 or 0.85 μM, there was a receptor reserve for taprostene at low agonist concentrations that declined incrementally with response and was essentially depleted at the Emax. These data are consistent with the ability of RO1138452 to depress the taprostene Emax at concentrations as low as 10 nM. Stated differently, occupancy of all available IP-receptor is required for taprostene to elicit its maximum response.
Conclusions. Herein, we report that human airway epithelial cells express functional IP-receptors. This finding adds to an expanding literature that documents the ubiquitous expression of IP-receptors in the lung and where, in vivo, selective agonists may have anti-inflammatory activity, antiviral activity, or both. Although taprostene incompletely suppressed chemokine output, greater inhibition can, theoretically, be achieved. Indeed, taprostene was a partial agonist, indicating that the IP-receptor system in airway epithelia can be activated further with ligands of higher efficacy. Furthermore, because cAMP inhibits the release of many mediators implicated in airway inflammation, selective agonists may have wide-spread activity on those proinflammatory and immune cells that express the IP-receptor subtype.
Our investigation also suggest that RO1138452 is a pseudo-irreversible orthosteric IP-receptor antagonist. The unexpected deviation from surmountable competitive behavior confirms the need, in drug discovery, to screen potential new medicines in the target tissue(s) of the relevant species as differences in receptor reserve and tissue phenotypes could have profound pharmacological and therapeutic implications.
Acknowledgments
We thank Merck Frosst for supplying L-161,982 and L-798,106 and Dr. Robert Newton (Department of Cell Biology and Anatomy, University of Calgary, Calgary, AB, Canada) for donating the CRE-reporter constructs.
Footnotes
-
M.A.G. is an Alberta Heritage Foundation for Medical Research Senior Scholar and is funded by the Canadian Institutes of Health Research (CIHR). D.P. is the recipient of a Canada Research Chair in Inflammatory Airway Diseases and is supported by the CIHR. S.L.T. acknowledges Nycomed Canada for postdoctoral fellowship support.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.107.129312.
-
ABBREVIATIONS: PGI2, prostacyclin; GPCR, G protein-coupled receptor; IP, prostacyclin; RO1138452, 4,5-dihydro-1H-imidazol-2-yl)-[4-(4-isopropoxybenzyl)-phenyl]-amine; COPD, chronic obstructive pulmonary disease; CXCL9, monokine induced by interferon-γ; CXCL10, interferon-γ inducible protein of 10 kDa; CRE, cAMP-response element; KSFM, keratinocyte serum-free medium; HAEC, human primary airway epithelial cell; IFN, interferon; ELISA, enzyme-linked immunosorbent assay; MOI, multiplicity of infection; PKA, cAMP-dependent protein kinase; PKI, cAMP-dependent protein kinase inhibitor; CMV, cytomegalovirus; BWA 868C, 3-benzyl-5-(6-carboxyhexyl)-1-(2-cyclohexyl-2-hydroxyethylamino)hydantoin; L-161,982, [4′-[3-butyl-5-oxo-1-(2-trifluoromethyl-phenyl)-1,5-dihydro-[1,2,4]triazol-4-ylmethyl]-biphenyl-2-sulfonic acid (3-methyl-thiophene-2-carbonyl)-amide]; L-798,106, 5-bromo-2-methoxy-N-[3-(2-naphthalen-2-ylmethyl phenyl)acryloyl]-benzene sulfonamide; Br, bromo; iloprost, (5E)-5-[(3aS,4S,5R,6aS)-5-hydroxy-4-[(E,3S)-3-hydroxy-4-methyl-oct-1-en-6-ynyl]-3,3a,4,5,6,6a-hexahydro-1H-pentalen-2-ylidene]pentanoic acid; taprostene, 3-[(Z)-[(1S,3R,4S,5R)-4-[(E,3S)-3-cyclohexyl-3-hydroxy-prop-1-enyl]-3-hydroxy-8-oxabicyclo[3.3.0]oct-7-ylidene]methyl]benzoic acid; DP, PGD2 receptor; EP, PGE2 receptor; TP, thromboxane receptor; AFP-07, 18,19-didehydro-7,7-difluro-16S,20-dimethyl-PGI2; TEI-9063, 17,20-dimethylisocarbacyclin.
-
↵
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material. - Received July 27, 2007.
- Accepted October 24, 2007.

- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}