





Abstract
Dehydroepiandrosterone (DHEA) prevents chronic hypoxia-induced pulmonary hypertension and associated right ventricle dysfunction in rats. In this animal model, reoxygenation following hypoxia reverses pulmonary hypertension but not right ventricle dysfunction. We thus studied the effect of DHEA on the right ventricle after reoxygenation, i.e. after a normoxic recovery phase secondary to chronic hypoxia in rats.
Right ventricle function was assessed in vivo by Doppler echocardiography and in vitro by the isolated perfused heart technique in three groups of animals: control, recovery (21 days of hypoxia followed by 21 days of normoxia) and recovery DHEA (30 mg·kg−1 every 2 days during the recovery phase). Right ventricle tissue was assessed by optical and electron microscopy.
DHEA abolished right ventricle diastolic dysfunction, as the echographic E wave remained close to that of controls (mean±sd 76.5±2.4 and 79.7±1.7 cm·s−1, respectively), whereas it was diminished to 40.3±3.7 in the recovery group. DHEA also abolished right ventricle systolic dysfunction, as shown by the inhibition of the increase in the slope of the pressure–volume curve in isolated heart. The DHEA effect was related to cardiac myocytes proliferation.
In conclusion, DHEA prevents right ventricle dysfunction in this animal model by preventing cardiomyocyte alteration.
Typical chronic lung hypoxaemic diseases, such as chronic obstructive pulmonary disease (COPD) [1], can lead to pulmonary hypertension (PH) and ultimately to right ventricular failure [2–4]. Rodents exposed to chronic hypoxia, either normo- or hyperbaric, consistute a classical animal model used to investigate mechanisms as well as therapeutic targets in this pulmonary vascular disease [5, 6]. In a rat model of hypobaric chronic hypoxia, we have previously demonstrated that dehydroepiandrosterone (DHEA), an adrenal steroid, prevents and decreases hypoxic PH and associated right ventricle hypertrophy [7]. In the same animal model, reoxygenation following hypoxia, i.e. a normoxic recovery phase of 21 days of normoxia secondary to a chronic hypoxic period of 21 days, also reverses PH as it normalises pulmonary pressure and antagonises vascular remodelling [8]. However, such normoxic recovery period does not correct right ventricular dysfunction [9]. We believe that such an experimental model may be relevant in patients suffering from COPD, as they may alternate between successive severe hypoxic episodes related to exacerbations [10] and fewer hypoxic episodes related their oxygen therapy [11].
As mentioned, DHEA has been studied in animal [7, 12] and human PH [13, 14]. More recently, it has been shown that DHEA can modulate cardiac function. DHEA reverses left ventricular stiffness and fibrosis, which typically accompanies ageing in mice [15], and decreases the production of type I collagen by cardiac fibroblasts [16]. DHEA can also modulate oxidative stress in the rat heart [17].
The purpose of this study was thus to evaluate the effect of DHEA on the right ventricle in this chronic hypoxia model followed by reoxygenation, i.e. followed by a normoxic recovery phase in rats. We examined the additional effect of DHEA on reoxygenation of the right ventricular structure and function both in vivo and ex vivo. The signalling pathway activated by DHEA was also determined.
METHODS
Animal model and DHEA treatment
The investigation was carried out in agreement with the Guide for the Care and Use of Laboratory Animals [18] and European Directives [19]. Adult male Wistar rats (220–240 g) were randomised into three groups. The first group of rats was kept in a hypobaric chamber for 21 days in order to induce a chronic hypoxic PH as previously described [7], followed by normoxia for 21 days (recovery group); the second group of rats was also kept in a hypobaric chamber for 21 days followed by normoxia for 21 days while being treated with DHEA (30 mg·kg−1 every 2 days) during the recovery period (recovery–DHEA group). These two groups were compared with the control group, which was kept at normal atmospheric pressure.
Assessment of heart tissue weight ratio and echocardiography
The right ventricle hypertrophy index was calculated as the ratio of the weight of the right ventricle to that of the left ventricle plus the septum; the right ventricle mass index was calculated as the ratio of right ventricle weight to bodyweight as previously described [20]. Echocardiography Doppler imaging was performed as previously described [9]. The right ventricle shortening fraction was then estimated as ((end-diastolic diameter - end-systolic diameter)×100)/end-diastolic diameter. The right ventricle diastolic function was estimated by the E-wave and the E/A peak velocity ratio. Pulse-wave Doppler (A and E wave) of the tricuspid valve was recorded in an apical four-chamber view.
Isolated and perfused heart technique
The isolated and perfused heart technique was performed as previously described [9].
Histological measurements
Optical and electronmicroscopy were performed as previously described [7]. The right ventricular tissue was fixed in 4% (w/v) formaldehyde and 3-μm thick sections were stained with haematoxylin, eosin and saffron. The number (N) of myocytes per ventricle was estimated according the following equation: N=(myocyte volume fraction×ventricular volume)/median myocyte volume [21]. The myocyte volume fraction used was 75%, and ventricular volume was calculated as the ventricular weight divided by the ventricular specific gravity (1.06 g·cm−3).
Reactive oxygen species measurement
Reactive oxygen species were measured by means of the electron paramagnetic resonance technique on right ventricle tissue, and was performed as previously described [22].
Mitochondrial activity assays
Complex 1 and citrate synthase activity assays were performed on right ventricle tissue as previously described [23].
Western blotting
Western blotting was performed on right ventricle tissue as described previously [23]. Antibodies against respiratory chain complex III core 2 and F1F0 adenosine triphosphate (ATP) synthase were obtained from Mitoscience (Eugene, OR, USA). The caspase 3, peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α), manganese superoxide dismutase (MnSOD) and endothelial nitric oxide synthase (eNOS) antibodies were obtained from Santa Cruz Biothechnology Inc. (Santa Cruz, CA, USA). The antiphospho-cyclic adenosine monophosphate response element binding (CREB) and antiproliferating cell nuclear antigen (PCNA) were obtained from Abcam Biochemicals (Bristol, UK).
Cell proliferation and apoptosis
Tissues were fixed in phosphate-buffered 4% formaldehyde and paraffin embedded. Myocyte cross-sectional areas were measured using an image-based quantitative analysis system (NanoZoomzer Digital Pathology Image software; Hamamatsu Photonics France, Massy, France). The outlines of 50 myocytes were traced in each animal studied. Cell proliferation and apoptosis detection were performed as previously described [24]. Proliferating cardiomyocytes were identified on the basis of PCNA co-immunostaining with desmin. The anti-PCNA monoclonal antibody was obtained from Calbiochem/Merck (Darmstadt, Germany). Apoptotic cardiomyocytes were detected using the ApopTag peroxidase kit (Millipore, Billerica, MA, USA) according to the manufacturer's instructions. Quantitative morphology was blindly performed on coded specimens. The tissue sections were scanned through systematic random field sampling using 0.143-mm2 calibrated fields captured with a bright-field microscope and digital image acquisition system (Olympus, Tokyo, Japan). 11–24 (mean±sem 18.1±1.2) fields per specimen were sampled through the complete tissue sections, and the numbers of PCNA+ cell nuclei were referenced to the sampled surface area. Lung tissue sections from a murine asthma model published elsewhere [25] were entered in the staining batches as terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labelling (TUNEL)+ control specimens.
Data analysis
Data values are expressed as the mean±sem. Statistical analyses were performed using the NCSS 5.0 software (NCSS, Kaysville, UT, USA), while intergroup differences were assessed by a Kruskal–Wallis ANOVA, as appropriate. In experiments with comparison of two conditions, as unpaired Mann–Whitney test was used. n refers to the animal sample size in the relevant experiment. Differences in the data were considered significant when p<0.05.
RESULTS
DHEA improves right ventricular dysfunction secondary to the normoxic recovery period
In vivo, the right ventricular systolic (shortening fraction) and diastolic (tricuspid E and A waves) functions were assessed by Doppler echocardiography (n=5 for each condition). Under basal conditions, there was no difference in the right ventricular shortening fraction between groups. The E wave was significantly decreased in the recovery group (40.3±3.7 versus 79.7±1.7 cm·s−1, p<0.001) and this effect was significantly prevented by DHEA treatment (76.5±2.4 cm·s−1, p<0.001) (fig. 1a). Similarly, the E/A wave was significantly increased in the recovery group (1.3±0.2 versus 0.4±0.01, p<0.001) and this effect was significantly prevented by DHEA treatment (0.4±0.08, p<0.001) (fig. 1b).

Dehydroepiandrosterone (DHEA) improves right ventricle dysfunction observed following a normoxic recovery phase secondary to chronic hypoxia. DHEA (30 mg·kg−1 every 2 days) was started during the normoxic recovery phase and right ventricle function was evaluated in vivo by Doppler echocardiography and ex vivo by the isolated perfused heart technique (n=5 per condition). The in vivo right ventricle diastolic function evaluated by a) the E wave and b) the E/A ratio was altered in the recovery group; this alteration was prevented by DHEA treatment. The ex vivo evaluation of the right ventricle c) systolic and d) diastolic function showed a positive effect of DHEA on the right ventricle systolic dysfunction (systolic pressure corresponding to the diastolic volumes: 80, 140, 160 and 180 mL) and right ventricle diastolic dysfunction (diastolic pressure corresponding to the diastolic volumes: 80–180 mL) in the recovery–DHEA group compared with the recovery group. *: p<0.05; ***: p<0.001.
Ex vivo, the right ventricular systolic and diastolic performances were further evaluated in the absence of right ventricular post load using the isolated perfused heart technique (n=5 for each condition). The recovery condition increased the systolic pressure for large diastolic volumes (80, 140, 160 and 180 mL; p<0.001) (fig. 1c) and this effect was significantly decreased by DHEA treatment. The recovery condition also increased the diastolic pressure for large diastolic volume (80–180 mL, p<0.001) (fig. 1d) and this effect was also decreased by DHEA.
DHEA improves right ventricle myocyte density during the normoxic recovery period by stimulating cardiomyocyte proliferation
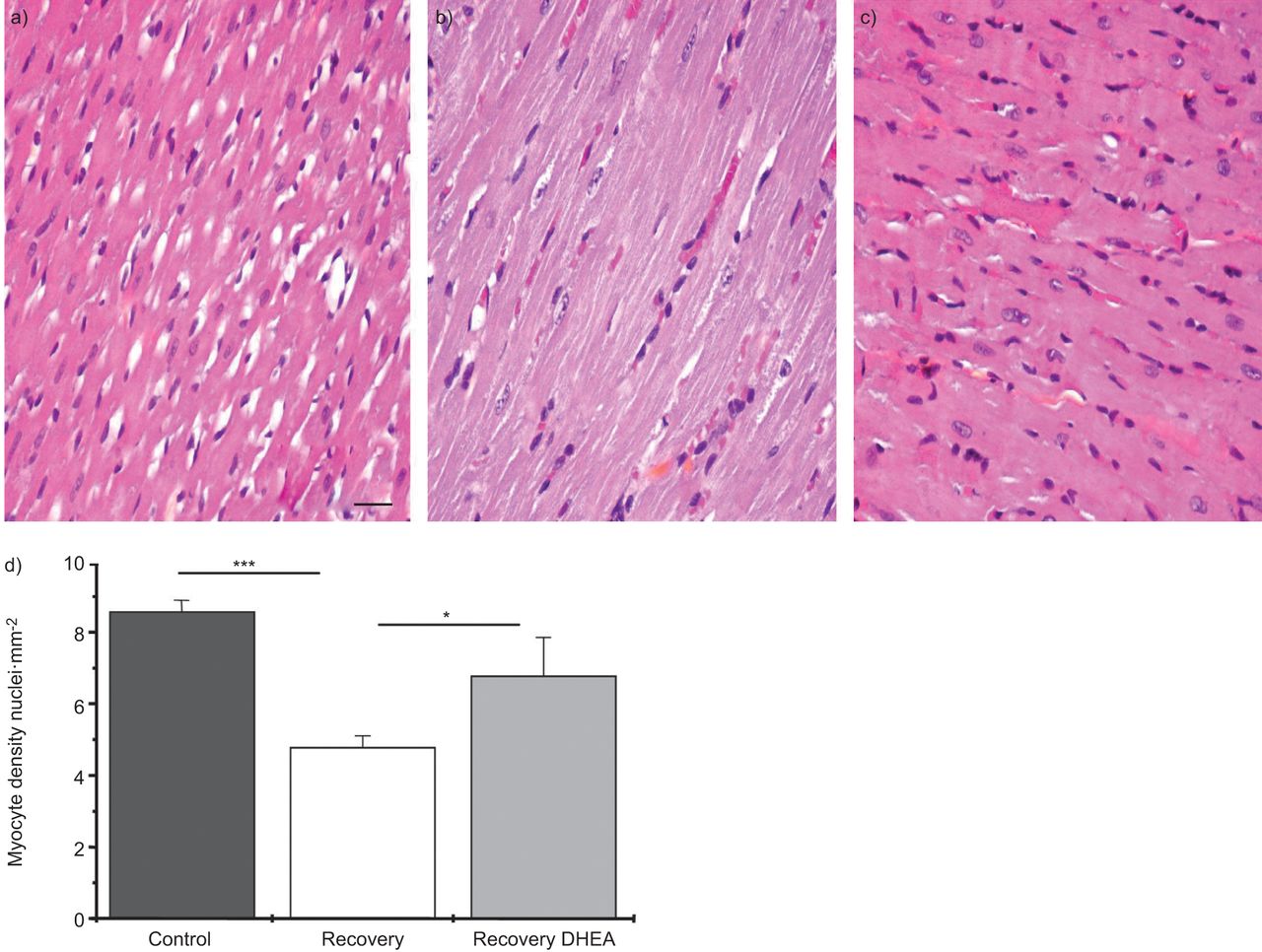
The right ventricle myocyte density (assessed as the number of nuclei per mm2) was studied by optical microscopy (nine slides per rat). The mean myocyte density was lower in the recovery group than in the control group (4.77±0.33 versus 8.56±0.32, p<0.001) and this effect was significantly prevented by DHEA treatment (6.77±1.08, p<0.05) (fig. 2). An estimation of the total number of myocytes was also determined and showed the same differences between groups (table 1). In order to determine whether the decrease in myocytes density was related to a loss of myocytes or myocyte hypertrophy, the right ventricle hypertrophy index, the right ventricle mass index and the myocyte cross-sectional area were determined. There was no difference in either index between the recovery and the recovery–DHEA groups (table 1). Moreover there was no difference in the myocyte cross-sectional area between groups (table 1). Therefore, the decrease in myocyte density was related to a loss of myocytes.

Dehydroepiandrosterone (DHEA) increases right ventricle myocytes density observed following a normoxic recovery phase secondary to chronic hypoxia. DHEA (30 mg·kg−1 every 2 days) was started during normoxic recovery phase and myocyte density was evaluated by optical histology (nine slides per rat). It showed a decrease in myocyte density secondary to the recovery period, which was significantly improved by DHEA treatment. Typical right ventricle tissue section for each a) control, b) recovery and c) recovery–DHEA group. Scale bar=50 μm. d) Summary data for myocyte density expressed as the number of nuclei per mm2. *: p<0.05; ***: p<0.001.
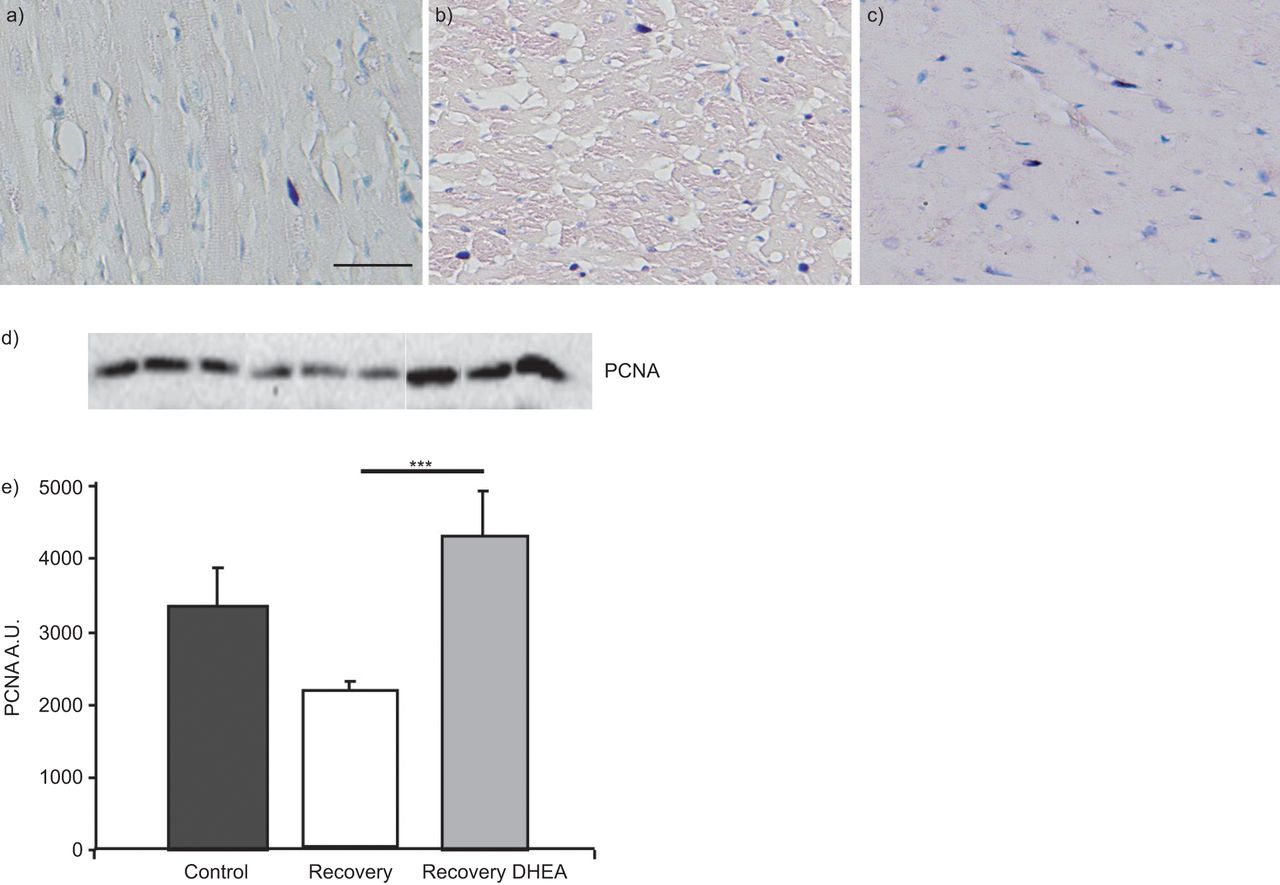
To examine the protective effect of DHEA, cell apoptosis was studied by the TUNEL+ staining technique and by Western blotting (cleavage of caspase 3). Representative fields of TUNEL+ cells were extremely rare, with no difference between the groups; the Western blot showed no significant difference when comparing caspase 3 activation. The PCNA staining (fig. 3a–c) showed a significant increase of in the recovery–DHEA group (8.6±1.6 cells·mm−3) compared with the recovery group (12.1±1.8 cells·mm−3, p<0.05) and this was confirmed by Western blot (fig. 3d).

Dehydroepiandrosterone (DHEA) increases right ventricle myocyte density observed following a normoxic recovery phase secondary to chronic hypoxia by stimulating cardiomyocyte proliferation. DHEA (30 mg·kg−1 every 2 days) was started during the normoxic recovery phase. Myocyte proliferation was evaluated by proliferating cell nuclear antigen (PCNA) co-immunostaining with desmin staining for a) control, b) recovery and c) recovery–DHEA groups and by d) Western blotting of PCNA, which was significantly improved with DHEA treatment. Scale bar=50 μm. d) A typical Western blot (performed in triplicate) for each group and e) summary data. A.U.: arbitrary units. ***: p<0.001.
DHEA prevents mitochondrial fragmentation induced by normoxic recovery
In a first set of experiments, the effect of normoxic recovery and DHEA treatment on: 1) the number of mitochondrial sections per cell area; 2) the mitochondrial section area; and 3) the matrix density. Sections were observed at a magnification of 6,000× in a series of randomly selected tissue sections (nine slides per rat). The number of mitochondrial sections was higher in the recovery group (4.07±0.20 versus 2.52±0.20 sections·μm-2; p<0.001) and DHEA significantly reduced this effect (2.94±0.10 sections·μm-2; p<0.001) (fig. 4a–g). There was no difference in the total mitochondrial section area between the groups. Accordingly, the mean mitochondrial section surface area was lower in the recovery group (0.099±0.004 versus 0.195±0.012 μm2; p<0.001) and DHEA prevented this effect (0.148±0.003 μm2; p<0.05) (fig. 4h). In a second set of experiments, mitochondrial ultrastructural abnormalities including partial cristolysis, disorganised cristae and matrix inclusions were observed at high magnification (20,500×). Abnormalities appeared in the recovery group and were less prominent after DHEA treatment (fig. 4d–f). The matrix density was also increased following treatment with DHEA as compared with the recovery group (matrix density 216±85% in the recovery group, p<0.05). This suggests a fragmentation of the mitochondrial network during recovery, which was prevented by DHEA.
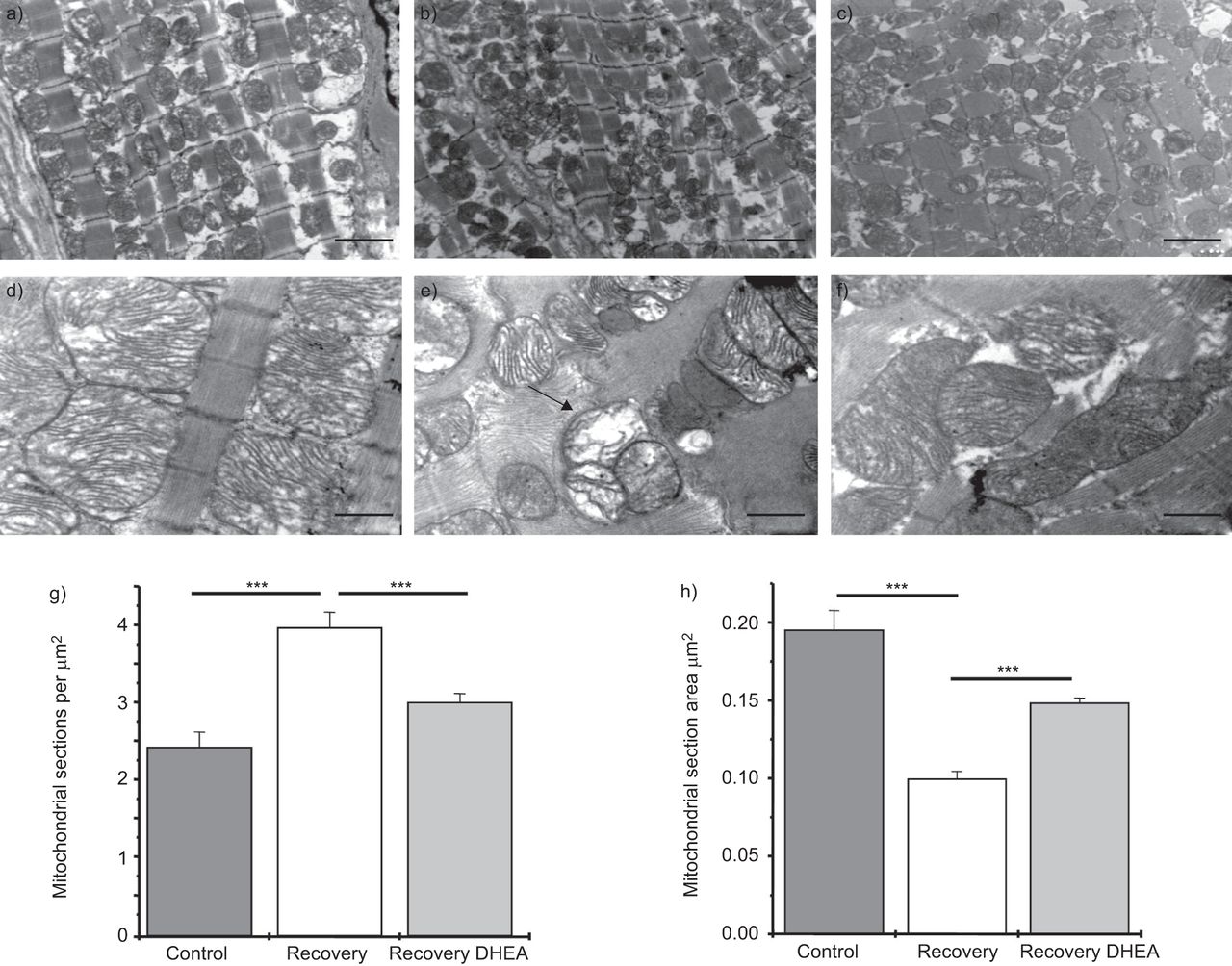
Dehydroepiandrosterone (DHEA) prevents mitochondrial fragmentation observed following a normoxic recovery phase secondary to chronic hypoxia. DHEA (30 mg·kg−1 every 2 days) was started during the normoxic recovery phase and mitochondrial network was evaluated by electron microscopy (a–c; magnification 6,000×, nine slides per rat). It showed a mitochondrial fragmentation (increase in mitochondrial sections (a–c, g) and decrease in mitochondrial section area (h)) in the recovery group; this fragmentation was significantly improved by DHEA treatment. Electron micrograph evaluation at larger magnification (d–f; 20,500×) showed mitochondrial ultrastructural abnormalities, including partial cristolysis, disorganised cristae and matrix inclusions in the recovery group; the alterations appeared to be less prominent after treatment with DHEA during the recovery period. Scale bars=1 μm. ***: p<0.001.
DHEA protective mechanisms include stimulation of PGC1α and eNOS via CREB activation
There was a 35±7% increase in PGC1α with DHEA treatment compared with the level measured after the recovery phase (p<0.001) (fig. 5a). As it has been recently reported that the expression of PGC1α can be stimulated in conjunction with the upregulation of eNOS expression in the heart [26], we additionally measured eNOS expression level. As expected, a 53±17% increase in the expression level of eNOS was observed in the treated heart (p<0.001) (fig. 5b). The eNOS gene promoter is controlled by phospho-CREB and is thereby stimulated in adverse situations of increased energy demand [27]. We observed a strong (136±18%) and significant (p<0.001) increase in the phospho-CREB levels in the DHEA-treated heart samples (fig. 5c).

Dehydroepiandrosterone (DHEA) protective mechanisms include the stimulation of peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α), endothelial nitric oxide synthase (eNOS) and cyclic adenosine monophosphate response element binding (CREB). DHEA (30 mg·kg−1 every 2 days) was started during the normoxic recovery phase secondary to chronic hypoxia. Western blot evaluation of a) PGC1α showed an increase in expression after DHEA treatment (p<0.001), b) eNOS showed a decrease in expression in the recovery group that was prevented by DHEA treatment and c) phospho-CREB showed a decrease in expression in the recovery group that was prevented by DHEA treatment. Typical Western blots are shown in triplicate for each group a–c for each group. Graphs show summary data. A.U.: arbitrary units. *: p<0.05; ***: p<0.001.
DHEA improves mitochondrial respiratory chain activity and biogenesis
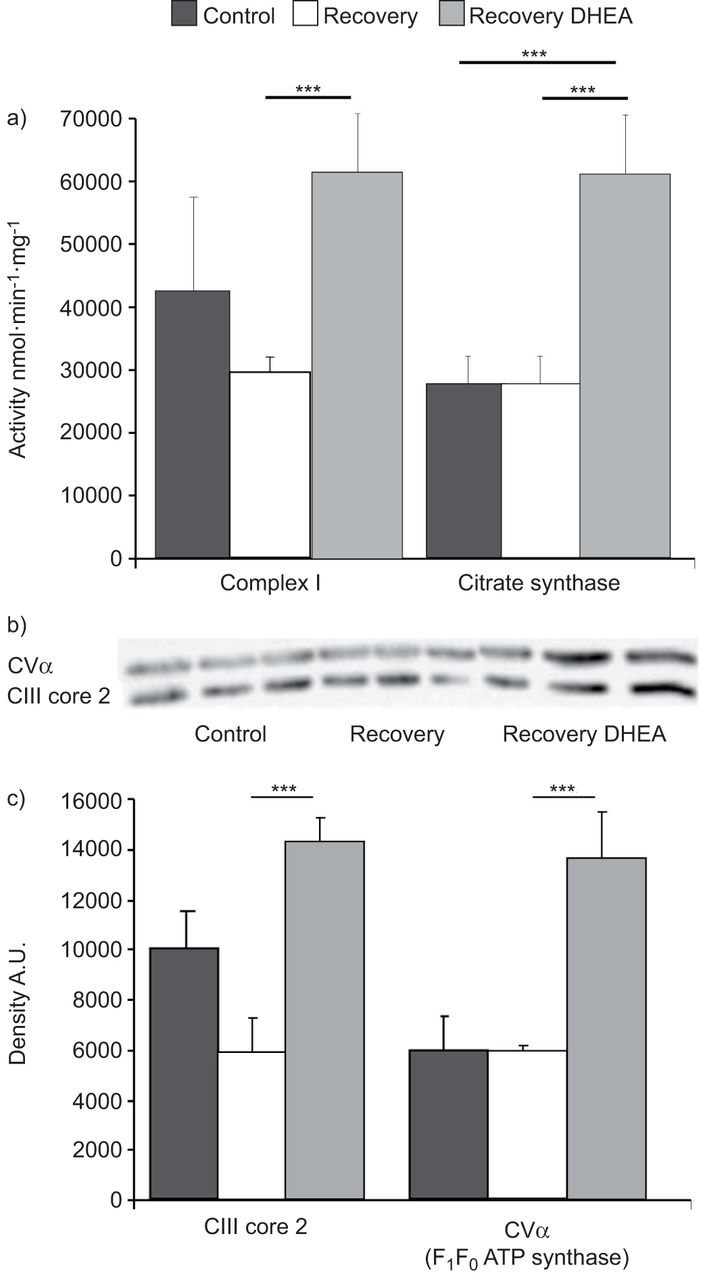
We measured the catalytic activity of complex I (reduced nicotinamide adenine dinucleotide–ubiquinone oxidoreductase) by spectrophotometry. We observed a large increase in complex I activity after the treatment with DHEA (p<0.001) (fig. 6a). We also measured the activity of citrate synthase that is typically proportional to the respiratory chain protein content [23]. Citrate synthase activity was increased by DHEA treatment to the same extent as complex I activity (p<0.001) (fig. 6a). The Western blot analysis of respiratory chain complex III and complex V (F1F0 ATP synthase) showed an increase in the content of these proteins (by a factor of two, following exposure to DHEA (p<0.001) (fig. 6b). These results suggest that DHEA activates mitochondrial respiratory chain protein expression.

Dehydroepiandrosterone (DHEA) improves mitochondrial respiratory chain activity and organellar biogenesis. DHEA (30 mg·kg−1 every 2 days) was started during the normoxic recovery phase secondary to chronic hypoxia. The mitochondrial respiratory chain activity was evaluated by spectrophotometry and the biogenesis by Western blotting. a) The catalytic activity of complex I (reduced nicotinamide adenine dinucleotide–ubiquinone oxidoreductase) and citrate synthase, measured by spectrophotometry), were largely increased after treatment with DHEA. b, c) Western blot evaluation of respiratory chain complex III and complex V (F1F0–ATPsynthase) showed a large increase in protein expression after DHEA treatment. b) A typical Western blot in triplicate for each group; c) summary data. A.U.: arbitrary units; CIII: complex III; CV: complex V. ***: p<0.001.
DHEA decreases cellular production of the superoxide anion and stimulates mitochondrial antioxidant defence
The production of the superoxide anion (O2-) was slightly increased, although not significantly, in the recovery group in comparison with the control group (371.9±48.3 A/(mg·mL−1) and 261.9±141.7 A/(mg·mL−1), respectively). DHEA significantly decreased O2- production (21.7±3.7 A/(mg·mL−1), p<0.001) (fig. 7a). We also measured the expression level of MnSOD that is typically upregulated under pathological conditions of oxidative stress [28]. Accordingly, our results indicate a significant increase in the MnSOD expression level following the recovery procedure with a significant decrease after DHEA treatment (p<0.001) (fig. 7b and c).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Dehydroepiandrosterone (DHEA) decreases cellular production of the superoxide anion (O2-) and stimulates mitochondrial antioxidant defences. DHEA (30 mg·kg−1 every 2 days) was started during the normoxic recovery phase secondary to chronic hypoxia. Production of superoxide anion was evaluated by the electron paramagnetic resonance (EPR) method. DHEA treatment induced a decrease in superoxide anion production. a) Data from EPR spectrometry are expressed as a ratio of the amplitude of the pic (A) out of the protein concentration of each pool of tissue in mg·mL−1. b, c) Mitochondrial antioxidant defences was assessed by b) Western blot evaluation of manganese superoxide dismutase (MnSOD) expression. It showed an increase in protein expression in the recovery group, which was significantly decreased after DHEA treatment. b) A typical Western blot (performed in triplicate) for each group; c) shows summary data. A.U.: arbitrary units. *: p<0.05; **: p<0.001.
DISCUSSION
In the present study, we have shown that DHEA improves both systolic and diastolic right ventricle dysfunction observed following a normoxic recovery phase secondary to chronic hypoxia in rats. This right ventricle dysfunction is associated with 1) a significant decrease in cardiac myocyte density and 2) mitochondrial fragmentation without alteration in the respiratory chain activity. DHEA prevented most of these observed modifications. Specifically, DHEA induced both an increase in mitochondrial energy protein expression and a decrease in O2- production, which was associated with an improvement in mitochondrial antioxidant defence and cardiomyocyte proliferation. DHEA treatment during the recovery had no effect on apoptosis, but stimulated cell proliferation. The signalling pathway of these effects involves an activation of the CREB protein leading to the subsequent expression of eNOS and stimulation of the regulator of mitochondrial biogenesis PGC1α. This suggests a novel mechanism by which DHEA partially antagonises heart alteration following a chronic hypoxia-recovery insult.
To the best of our knowledge, this is the first study exploring the effect of DHEA on right ventricle impairment induced by a normoxic recovery phase following chronic hypoxia exposure. We have previously shown that such a normoxic recovery phase secondary to chronic hypoxia induces nonreversible right ventricular dysfunction and dysplasia in rats [9]. Morphometric analyses demonstrated a mitochondrial network fragmentation that is typically observed in situations of mitochondrial dysfunction [29]. A cardiac dysfunction related to an alteration in mitochondrial structure and activity has been previously described in other models [30, 31]. Mitochondrial fragmentation could indicate a decrease in cellular ATP levels, as mitochondrial transition from the reticular to the fragmented state is dependent upon ATP levels. The observed decreased cardiac cell density in the right ventricle after the normoxic recovery phase could be explained by the onset of apoptosis during the chronic hypoxia phase [32]. However, such an effect was not observed after the normoxic recovery phase, probably because apoptotic cells are rapidly removed by the immune system.
In the present study, DHEA stimulated mitochondrial respiratory chain protein expression and this effect was associated with an improvement in cell density, which suggests that impairment in mitochondrial energy metabolism did limit cell growth in the recovery group. The alteration in the overall and internal structure of the mitochondria (i.e. fragmentation of the mitochondrial network and cristolysis) observed in the recovery group supports this hypothesis. In the DHEA-treated group, mitochondrial configuration returned to normal and oxidative phosphorylation proteins were upregulated. Taken together, these observations suggest that DHEA stimulates mitochondrial biogenesis in the right ventricle, as has previously been shown in the liver and the brain [33, 34].
Our study also highlights the cellular effect of DHEA and the associated signalling pathways in the right ventricle. PGC1α is a transcriptional co-activator implicated in mitochondrial biogenesis [35, 36] that can be stimulated in conjunction with the upregulation of eNOS expression in the heart [26]. The mechanism of such upregulation of eNOS may be due to the DHEA-induced increase in CREB, a factor controlling eNOS gene promoter. Taken together, these data indicate a possible implication of the eNOS/PGC1α/CREB pathway in the effects of DHEA. Interestingly, a recent study [37] indicates that PGC1α controls DHEA synthesis in the liver. Together with our observations, this could indicate that DHEA participates in feedback control of PGC1α expression or activation. A feedback control of PGC1α expression by its target genes would be a remarkable feature of the transcriptional network and regulatory signals that govern mitochondrial biogenesis. Furthermore, we show that DHEA decreases O2- production. This could be one of the mechanisms underlying the protective effect of DHEA. Indeed, reactive oxygen species are known to be involved in mitochondrial damage and cellular injury, and may lead to heart failure [2, 38]. Moreover, the MnSOD level was higher during the normoxic recovery phase (where oxidative stress was measured) and decreased with DHEA treatment. Our data therefore support previous observations that MnSOD expression is controlled by the reactive oxygen species concentration [39].
In conclusion, DHEA prevents right ventricle dysfunction, remodelling and myocyte alteration observed following a normoxic recovery phase secondary to chronic hypoxia. The stimulation of the antioxidant defence and of the mitochondrial metabolism via an activation of the eNOS/PGC1α/CREB axis may explain the decrease in reactive oxygen species production and the increase in cell proliferation observed after DHEA treatment. 1) Such an experimental model may be relevant in patients suffering from COPD and PH because they alternate between successive severe hypoxic episodes related to exacerbations and less hypoxic episodes related their oxygen therapy, and 2) DHEA is well tolerated [40] and is under current investigation in PH in humans [14]. Thus, DHEA may prove to be a valuable molecule that deserves further studies, particularly in PH secondary to COPD, where right ventricular failure is one of the main causes of mortality.
Acknowledgments
The authors want to thank A-M. Lomenech, P. Techoueyres, S. Vautrat, G. Simon, S. Guerit and L. Parios (all Inserm U1045, Bordeaux, France) for their technical assistance.
Footnotes
Statement of Interest
None declared.
- Received January 21, 2011.
- Accepted March 26, 2012.
- ©ERS 2012
REFERENCES










