





Abstract
Pulmonary hypertension is a hallmark of high-altitude pulmonary oedema (HAPE) and of congestive right heart failure in subacute mountain sickness (SMS) and chronic mountain sickness (CMS) in the Himalayas and in the end-stage of CMS (Monge's disease) in the Andes.
There are studies to suggest that transmission of excessively elevated pulmonary artery pressure and/or flow to the pulmonary capillaries leading to alveolar haemorrhage is the pathophysiological mechanism of HAPE.
In the Himalayas, HAPE was successfully prevented by extending the acclimatisation period from a few days to 5 weeks, however, this did not prevent the occurrence of congestive right heart failure after several weeks of stay at 6,000 m. This leads to the concept that rapid remodelling of the small precapillary arteries prevents HAPE but not the development of right heart failure in SMS and CMS.
Unresponsiveness of pulmonary hypertension to oxygen at high altitude and its complete resolution only after weeks of stay at low altitude suggest that structural rather than functional changes are its pathophysiological mechanism. Since pulmonary hypertension at high altitude is the driving force leading to high-altitude pulmonary oedema and “high-altitude right heart failure” in newcomers and residents of high altitude, the authors propose to adjust current terminology accordingly.
- chronic mountain sickness
- high-altitude pulmonary oedema
- Monge's disease
- pulmonary hypertension
- right heart failure
- subacute mountain sickness
The physiological response of the pulmonary circulation to hypobaric and normobaric hypoxia is to increase pulmonary arteriolar resistance. The magnitude of hypoxic pulmonary vasoconstriction is highly variable between humans, probably based on genetics and adaptive mechanisms. Sites of hypoxic pulmonary vasoconstriction are small pulmonary arterioles and veins of a diameter of <900 µm, the veins accounting for∼20% of the total increase in pulmonary vascular resistance caused by hypoxia 1, 2. The structural changes in small pulmonary arteries and veins appear to reflect this genetically based and adaptive process 3–5 in humans and animals. Excessive hypoxic pulmonary vasoconstriction, and thus susceptibility to develop a right heart failure within weeks at high altitude, was first described in Colorado cattle 6. Indian soldiers stationed at an altitude between 5,800–6,700 m 7 and Han infants in Lhasa 8 develop severe congestive right heart failure within weeks or months after arrival at high altitude. An excessively elevated pulmonary artery pressure (PAP) has not only been reported to cause high altitude cor pulmonale within weeks, months, or years in newcomers, but also in high-altitude residents of the Andes and nonacclimatised climbers prone to high-altitude pulmonary oedema (HAPE) 9. The results of these studies suggest that an excessive rise in PAP is a common denominator in HAPE 9, the syndrome described by Sui et al. 8 in infants and by Anand et al. 7 in adults at 6,700 m and termed “subacute mountain sickness” (SMS) of the infant and the adult, respectively, and in the illness of the high-altitude residents of the Andes termed “chronic mountain sickness” (CMS) or “Monge's disease” in its end-stage 10.
Unfortunately, the term “SMS” is misleading. In fact, this name was originally used by Monge 11 to describe persistent symptoms of acute mountain sickness, for weeks and months after ascent to high altitude. In the original description, these patients did not present with clinical signsofpulmonary hypertension or congestive heart failure. Similar confusion exists between the terms “Monge's disease” and “CMS”. The term “CMS” is used to describe both high-altitude excessive erythrocythosis in South Americans and congestive failure of the right heart without excessive polycythaemia in the Himalayas 12–14. Recently, Ge and Helun 13 proposed to change the term “CMS” to “high-altitude heart disease” for congestive right heart failure without excessive polycythaemia in the Himalayas 13.
The aim of this article is to highlight common clinical andpathophysiological aspects between acute, subacute andchronic high-altitude disease associated with pulmonary hypertension and, based on this common entity between them, propose a new possible classification and terminology.
Effects of acute exposure to high altitude
In healthy lowlanders at altitudes between 3,800–4,600 m, invasively assessed resting mean PAP ranged 15–35 mmHg (mean 25 mmHg) and the systolic PAP 27–48 mmHg (mean 37 mmHg) 15–17 (fig. 1⇓). At altitudes of >5,000 m, the mean PAP was assessed during a right heart catheterisation in sixresting healthy acclimatised volunteers during Operation Everest II 21. At a barometric pressure of 347 Torr (6,100 m) and at 282 Torr (7,620 m) the mean PAP was on average 19 mmHg and 34 mmHg, respectively. At both altitudes, physical exercise significantly increased mean PAP at 347 Torr on average to 41 mmHg and at 282 Torr to 54 mmHg. Among newcomers and visitors at high altitudes (>3,000 m), there are healthy individuals who present with excessive mean PAP values, which may exceed the 40 mmHg mark. In lowlanders susceptible to HAPE, mean PAP was on average 38 mmHg with a range of 31–51 mmHg 17. These results are consistent with an earlier report in five HAPE‐susceptible subjects that showed a mean PAP of 39 mmHg (range 22–47 mmHg) 24 h after arrival at 3,100 m 19. However, as shown in figure 1⇓ there is a considerable overlap between the mean PAP values reported in HAPE‐resistant and -susceptible individuals at high altitude. This observation may suggest that other mechanisms than pressure contribute to oedema of the lung in this acute setting.
Consistently, pulmonary heamodynamic measurements at rest performed in early HAPE 17 and in all patients admitted to the hospital with HAPE 22–27, show that left atrial pressure, as assessed by occluded (or wedged) PAP, right atrial pressure and cardiac output are normal in HAPE. Recently, using the method of arterial occlusion, which is likely to measure pressures in vessels close to 100 µm in diameter 28, it was demonstrated that the pulmonary capillary pressure (Pc) is elevated in HAPE. Pc was on average 16 mmHg (range 14–18 mmHg) in HAPE‐susceptible subjects without pulmonary oedema and 22 mmHg (range 20–26 mmHg) in those, who developed HAPE 17 (fig. 2⇓). These results suggest that the Pc threshold value for oedema formation in this setting is 20 mmHg. Since there is evidence that the small arterioles are the site of transvascular leakage in the presence of markedly increased PAP in hypoxia 29 and that pulmonary veins contract in response to hypoxia 30, 31 increasing the resistance downstream of the region of fluid filtration 32, it is likely that in the absence of altered pulmonary capillary permeability, elevated hydrostatic pressure in the pulmonary capillary plays an important role in the pathogenesis of HAPE. However, some element of regional heterogeneity of hypoxic pulmonary vasoconstriction (either at arterial and venous sites or both) are still necessary to explain the heterogeneity of regional oedema, at least as observed on chest radiographs or computed tomography scans 33.
The key role of elevated PAP in the pathogenesis of HAPE is demonstrated by the data showing that this condition is prevented or improved by the use of pulmonary vasodilators 34–36. Recent data showing that the inhalation of a β2‐agonist at a high dose during rapid exposure to 4,559 m prevented the development of HAPE, cannot be taken as an argument against the role of elevated capillary pressures in the pathogenesis of HAPE 37. In fact, it is likely that the improvement of alveolar transepithelial transport by β2‐agonists may shift the Pc threshold for alveolar flooding to higher Pc values by improving the equilibrium between the fluid moving across the blood-gas barrier. Moreover, although systolic PAP was not different between the placebo and the β2‐agonist-treated subjects in this study, it cannot be excluded that β2 stimulation at the level of the alveolar capillaries may decrease resistance in small pulmonary arteries or veins, or both.
Recent examinations of the content of alveoli in bronchoalveolar lavage (BAL) showed that both the subjects with HAPE and those who will develop HAPE within the next 24 h presented with elevated red blood cell (RBC) counts and serum-derived protein concentration, and normal alveolar macrophages and neutrophils counts and concentration of proinflammatory mediators in their BAL fluid 38. Interestingly, the albumin concentration and the number of the RBCs in the BAL fluid were significantly correlated with systolic PAP measured by echocardiography. The threshold for albumin at a systolic PAP was ∼40 mmHg and for RBCs ∼60 mmHg (fig. 3⇓).
In conclusion, all these recent results suggest that HAPE isahydrostatic type of pulmonary oedema; its pathophysiological mechanism is an excessive vasoconstriction of small pulmonary arteries and veins to hypoxia, which probably leads to an overdistension of the pulmonary capillaries, because of uneven distribution of blood flow and/or increased postcapillary resistance. This possibly leads to open cellular junctions and/or cause stress failure of the alveolo-capillary membrane. Recent BAL data suggest that signs of inflammation found in the BAL fluid of patients with advanced HAPEis a secondary event. Impairment of the alveolar transepithelial water transport and systemic inflammation may contribute to impaired fluid homeostasis across the blood-gas barrier.
Effects of subacute exposure to high altitude
Indirect evidence for elevated PAP has been found in infants of Han descent born at low altitude, who died after an average of 2 months of residence in Lhasa 8 and in Indian soldiers, who failed to acclimatise at the very high altitude of 5,800–6,700 m 7. In infants, autopsy revealed massive hypertrophy and dilatation of the right ventricle, dilatation of the pulmonary trunk, extreme medial hypertrophy of the muscular pulmonary arteries and muscularisation of the pulmonary arterioles 8. In Indian soldiers, clinical features compatible with an acute congestive right heart failure developed during weeks 3–22, on average 11 weeks after they were stationed at altitudes between 5,800–6,700 m 7. Before trekking to their post at extreme altitude, the soldiers had acclimatised during 1 week at 3,000 m and 1–3 weeks at altitudes between 3,000–4,500 m. After airlift to low altitude, clinical examination revealed tachypnoea, tachycardia, stasis of the jugular veins, enlargement of the liver and ascites. The electrocardiogram showed right axis deviation, right ventricular hypertrophy and T‐wave inversion V1 to V5–6. Chest radiograph revealed an enlargement of the heart, prominent vascular pedicules but no pulmonary infiltrates. Echocardiography confirmed the enlargement of the right ventricle and showed normal dimensions and ejection fraction of the left ventricle. On admission, mean PAP was on average 26 mmHg at rest and increased to 39 mmHg during mild exercise, the cardiac index increased from 3.15 to 5.28 L·min−1·m−2. After 12–16 weeks, mean PAP decreased on average to 16 mmHg and the cardiac index was 3.5 L·min−1·m−2. Pulmonary artery occluded pressure averaged 11 mmHg at rest, 13 mmHg during exercise and 8 mmHg after recovery. Characteristically, there was no excessive polycythaemia in these soldiers. The mean haemoglobin concentration was 18 g·dL−1 (range 14–23 g·dL−1) and the mean haematocrit was 61% (range 48–72%). In both infants and adults, right heart failure with signs of congestion developing within weeks or months of stay at high altitude has been called infantile and adult SMS, respectively.
The pathophysiological mechanism of SMS, which could also be named subacute high-altitude right heart failure, is incompletely understood. It is reasonable to assume that the initial stimulus leading to excessively elevated PAP is not different from HAPE. Since remodelling of the small weekly muscularised and nonmuscular pulmonary vessels may start within hours of exposure to hypoxia 39–41, it is conceivable that gradual ascent to high altitude may lead to a more homogeneous distribution of hypoxic pulmonary vasoconstriction, hence a better protection of the pulmonary capillaries from elevated pressure and high flow. However, thickening of the pulmonary artery media increases pulmonary vascular resistance, which in turn cause PAP to increase and the right ventricle to fail. Thus, it is possible that HAPE susceptible subjects after gradual ascent to high altitude and a stay there for weeks, months or years may also be prone to develop high-altitude right heart failure. Moreover, additional factors, such as increased neurohumoral activity and hypoxaemia due to subclinical pulmonary oedema following impaired diastolic left ventricular function, may also add to the pathogenesis of SMS 42.
Effects of chronic exposure to high altitude
Mean resting PAP has been reported to be lowest in Tibetans compared with Han Chinese high-altitude residents and South- and North-American natives. At similar altitudes between 3,658–3,950 m, mean PAP was on average 14 mmHg in Tibetans, 28 mmHg in Han Chinese residents of the Qinghai Province and 20 mmHg among natives of South America (fig. 1⇓). In Leadville, CO, USA, mean PAP in healthy males living at 3,100 m averaged 24 mmHg. Compared with Tibetans, North Americans have a greater rise in the PAP after hypoxic stimulus 4.
Transition from foetal to mature patterns of pulmonary circulation in newborns at high altitude, compared with infants at sea level, is slower and may even fail to develop. It has been reported that infants born at altitudes between 3,500–4,500 m may show persistent near systemic PAP values for some time after birth 43, and that in some cases elevated mean PAP (∼40 mmHg) persisted during infancy 44. These observations may be linked to right heart failure in infants after birth 8 and to chronic high-altitude pulmonary hypertension in adulthood 45.
The first symptoms associated with CMS are headache, dizziness, fatigue, insomnia and cognitive dysfunction: somnolence, slowed mental function, confusion and impaired memory. These symptoms suggest a loss of acclimatisation in these individuals born or living at high altitude for years. Episodes suggestive of right heart failure with dyspnoea, cough, turgid jugular veins and peripheral oedema follow the first symptoms within a few years in Han Chinese 13, 14, but are rare in residents of high altitude in the Andes 20. In both populations, marked cyanosis of the face and fingers, clubbing of the digits, hepatomegaly and ascites are present in the late stage of the disease. In Han Chinese and South Americans chest radiography shows enlargement of the heart, dilatation of the pulmonary trunk and general dilatation of the small lung vessels. Kearly's B lines are characteristically absent in these patients 14. These symptoms suggest that congestive right heart failure is associated with excessively elevated PAP in both populations. Pulmonary haemodynamic measurements were performed in a total of 16 natives of the Andes 20, most of them performed at an altitude of 4,300 m, and in the five Han Chinese, who developed the disease after 11–36 yrs of stay in Lhasa (3,658 m) 14. Mean PAP averaged 45 mmHg in South Americans and 40 mmHg in Han Chinese (fig. 1⇓). In all subjects, right atrial pressure, pulmonary artery occluded pressure (wedge pressure) and cardiac output was normal.
In natives of the Andes, CMS, also named Monge's disease, begins insidiously in adult life, often during the fourth decade, and is characteristically associated with excessive polycythaemia 11. Since there is evidence that chronic hypoxia irreversibly desensitises the reflex response to acute hypoxia mediated by peripheral chemoreceptors 46, 47, it has been suggested that hypoventilation leading to severe hypoxaemia, and hence to excessive erythrocytosis and in its end-stage, pulmonary hypertension, is the cause of Monge's disease. However, more recent studies, comparing the hypoxic ventilatory response (HVR) between patients with CMS and healthy natives of high altitude 48, 49, did not confirm this earlier conclusion 46, 47. Taken together, compared with sea-level residents, in these studies it has been found that both healthy resident of high altitude and patients with CMS present a decreased ventilatory response to hypoxia, the difference between those with and without CMS being not statistically significant 49. However, compared with healthy high-altitude residents, patients with CMS present higher end-tidal carbon dioxide tension and lower end-tidal oxygen tension, suggesting an HVR‐independent lower level of alveolar ventilationinthese subjects 47–49. Thus, from the results of all these studies, it may be concluded that a lower level of alveolar ventilation rather than a reduced peripheral chemosensitivity to hypoxia is the primary mechanism leading to hypoxaemia, hence excessive erythrocytosis, and hence pulmonary hypertension in patients with CMS or Monge's disease.
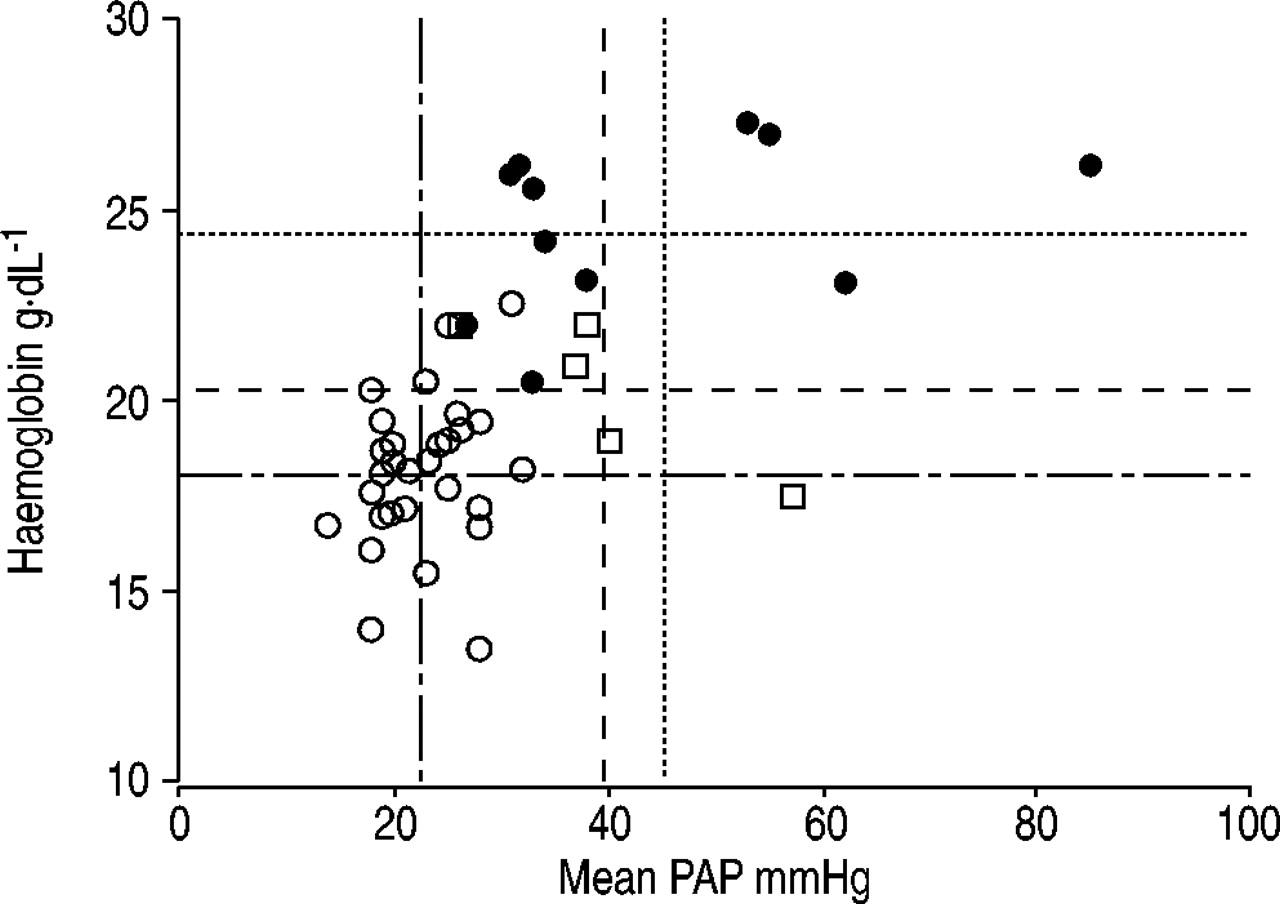
In subjects with Monge's disease, the haematocrit value on average exceeds the 70% mark (range 65–85%), and at an altitude of 4,000–4,500 m the arterial oxygen saturation (Sa,O2) is <81%. In contrast, healthy residents living at an altitude of 4,000–4,500 m have a haematocrit of ∼55–60% and an Sa,O2 of >81% 11, 50, 51. It is reasonable to assume that in CMS both excessive erythrocytosis and low haemoglobin oxygen content contribute to high-altitude pulmonary hypertension. However, there are cases of CMS with excessive polycythaemia but without pulmonary hypertension 52. Figure 4⇓ summarises haemoglobin and mean PAP pairs published in Han Chinese 14, and South Americans without 18 and with CMS 20. The figure shows that for the pooled data (n=47), there is a significant correlation of the second order between haemoglobin and PAP (r=0.64, p<0.001), but that, as previously reported 20, 53, this relationship is lost in patients with CMS (r=0.25, p=0.62). Furthermore, the figure illustrates that at a comparable mean PAP of ∼40 mmHg (mean±sd: Han Chinese 40±11 versus South Americans 45±18, p=0.87), haemoglobin concentration is lower in Han Chinese (mean±sd 20.3±1.9 g·dL−1, range 17–22) than in the natives of the Andes (mean±sd 24.6±2.1 g·dL−1, range 20–27; p<0.01, Mann-Whitney U‐test). The haemoglobin concentration in healthy individuals in Lhasa (3,600 m) is ∼17 g·dL−1 14 and in La Oroya in Peru (3,690 m) ∼18 g·dL−1 18. Thus, these results suggest that excessive hypoxic pulmonary vasoconstriction rather than polycythaemia-associated increased blood viscosity is the predominant cause of right heart failure in high-altitude residents with CMS.
High-altitude pulmonary hypertension
Pulmonary haemodynamic measurements performed in children and young adults show persistence of elevated PAPat high altitude for weeks, months or years 44, 54. Histological examination of the pulmonary vessels in high-altitude residents, who died from causes other than CMS, show persistence of the typical foetal patterns (thickened media) 45, 55. The experience of the Indian Army that its soldiers, if airlifted to extreme altitudes developed HAPE in up to 15% 56, and if acclimatised during several weeks before the stay at extreme altitude, develop congestive failure of the right heart, is highly suggestive for persistently elevated PAP once exposed to high altitude. Successful prevention of HAPE with better acclimatisation, in spite of probable excessively elevated PAP, suggests a remodelling process of the pulmonary precapillary vessels that protect the capillaries from exposure to high hydrostatic pressure or blood flow. The observation that highaltitude-induced changes of the pulmonary vasculature and right heart are reversible once moving to low altitude andthat some high-altitude residents when returning to highaltitude develop HAPE 57, are further arguments in favour of a rapid remodelling process of the pulmonary vessels in response to changes in air oxygen content. Moreover, the evidence that at high altitude, neither in acclimatised healthy subjects 21, HAPE‐susceptible subjects 17 norin high-altitude residents 58, supplemental oxygen significantly decreases PAP is also in line with this remodelling concept.
In rats exposed to hypoxia, precapillary vessels of a diameter of ∼25 µm, which normally do not have smooth muscle cells, begin to generate from adventitial fibroblasts within 24 h 39. Light microscopic examination of nonmuscular arterioles after exposure to hypoxia show that smooth muscle began to appear by day 2 at simulated altitude, the proportion of muscularised arterioles increasing along with increasing PAP 40, 41. Interestingly, all these studies show that after return to normoxia, smooth muscle cells persisted in normally nonmuscularised arterioles, suggesting that smooth muscle cells may remain for a very long time after chronic exposure to hypoxia. If applicable to humans, these findings suggest that hypoxia-associated smooth muscle proliferation in originally weakly muscularised arterioles and normally nonmuscular pulmonary vessels is likely to be the pathophysiological mechanism of high-altitude pulmonary hypertension, leading finally to high-altitude right heart failure either in SMS or CMS. Conversely, structural remodelling of the precapillary pulmonary vessels also may be crucial for the protection of the pulmonary capillaries from excessively elevated PAP, flow or both. Thus, if it is postulated that heterogeneity of hypoxic vasoconstriction is at the origin of HAPE, it is imaginable that the generation of new smooth muscle cells contributes to homogenise the vasoconstrictor response to hypoxia, protecting pulmonary capillaries from overperfusion, and hence alveoli from fluid flooding. Taken together, it cannot be excluded that individuals who present with high-altitude pulmonary hypertension may share common, at the present time unknown, genes that influence the magnitude of the pulmonary arterioles and/or veins to respond to hypoxia and the generation of smooth muscle cells from adventitial fibroblasts in weakly and nonmuscularised pulmonary vessels.
Terminology
Acute exposure to high altitude leads to acute mountain sickness and HAPE, two conditions with different pathophysiological mechanisms. In this setting, HAPE is indisputably the condition that is associated with high-altitude pulmonary hypertension. Unfortunately, terminology used todescribe high-altitude diseases associated with subacute and chronic exposure to environmental hypoxia is misleading. However, if the concept that high-altitude right heart failureisthe consequence of pulmonary hypertension in this setting is accepted, subacute and chronic high-altitude diseases could easily be classified in those with and without high-altitude pulmonary hypertension, hence cardiac failure (table 1⇓).
Monge 11 originally used the name SMS to describe the persistence of headache, anorexia, nausea, dizziness and difficulty to sleep, usual symptoms of acute mountain sickness, during weeks and months after arrival at high altitude. Typically, in all these patients, mainly mining workers of the Peruvian Andes, physical findings suggesting congestive failure of the right heart were absent. Unfortunately, the name “SMS” has also recently been used to describe the rapid (within a few weeks or months after ascentto high altitude) development of congestive right heartfailure in Han Chinese infants and Indian soldiers 7,8.According to the authors concept, the name “SMS” should be reserved for the description of the original syndrome published in 1937, and the name “cardiac SMS” should be used to describe the syndrome described by Anand et al. 7.
Monge and Whittembury 10 originally described chronic mountain sickness as a syndrome characterised bytriad excessive erythrocytosis, severe hypoxaemia and impaired mental function. Right heart failure was not included in the triad, because in the Andes it is a rare complication in the end-stage of the disease. Therefore, inanalogy to the condition of subacute right heart failure, theauthors propose to name the condition of congestive rightheart failure without excessive erythrocytosis, as it is mainly observed in immigrants born at low altitude after years of residence at high altitude, “cardiac chronic mountain sickness”. Consequently, the names “chronic mountain sickness” and “Monge's disease” should be reserved to describe chronic high-altitude disease associated with excessive erythrocytosis, severe hypoxaemia and hypercapnia at thegiven altitude preceding the development of a congestive failure of the right heart in its end-stage. Thus, in its early stage Monge's disease could be ascribed as a diseaseofrespiratory type (elevated end-tidal carbon dioxide tension), when alveolar hypoventilation is predominant, and one of cardiac type (right heart failure), when cardiac congestion is predominant in its end-stage.

Individual invasive measurements of mean pulmonary artery pressure (PAP) in sea-level (SL) and high-altitude residents obtained in the Alps, the Himalayas and in South America. Open circles show values measured in healthy lowlanders (LL) and healthy residents of high altitude. In healthy LL, measurements were performed at SL 17, 18 and at altitudes between 3741–4559 m, respectively 15–17. In residents of high altitude living in Tibet 4 and in the central Andes of Peru 18, PAP was measured at 3658 m, 3690 m and 4260 m, respectively. Open squares (high-altitude pulmonary oedema (HAPE) prone) show mean PAP values obtained at altitudes between 2600–4559 m in nonacclimatised HAPE‐susceptible subjects without HAPE (No HAPE; 3100–4559 m) 17, 19, with HAPE 17 in an early stage (Early HAPE; 4559 m), and with HAPE in an advanced stage (Adv HAPE; 2600–4331 m). In the latter group, mean PAP was measured at hospital admission. Open triangles show mean PAP values measured in high-altitude residents with chronic mountain sickness (CMS) and clinical signs of congestive right heart failure in the Himalayas (Han Chinese; HC) and the Andes 14, 20. The horizontal bars indicate the median mean PAP for each group of subjects. The figure illustrates that in ∼50% of the subjects with HAPE and CMS, mean PAP exceeded the 40 mmHg mark.

Relationship between individual pulmonary capillary pressure (Pc) and mean pulmonary artery pressure (PAP) assessed using the arterial occlusion technique, in controls (○), high-altitude pulmonary oedema (HAPE)‐susceptible subjects without (□) and with (•) pulmonary oedema 17. The figure shows that there is a good correlation between Pc and mean PAP, and that in all subjects who develop HAPE, Pc was higher than 19 mmHg. However, a cut-off for mean PAP could not be found.

Individual bronchoalveolar lavage (BAL) red blood cells (RBCs; •) and albumin (○) concentration plotted against systolic pulmonary artery pressure (PAP) at high altitude (4559 m). The figure shows that the threshold systolic PAP for the appearance of albumin in the BAL fluid was 35 mmHg and for RBCs >60 mmHg 38.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Relationship between haemoglobin and mean pulmonary artery pressure (PAP) in 30 healthy high-altitude residents 18 (○), five Han Chinese 14 (□) and 12 South Americans 20 (•) with chronic mountain sickness (CMS). Lines indicate individual population mean values for the haemoglobin and the mean PAP. ═: mean CMS Andes; - - -: mean CMS Himalayas; –– ‐: mean non‐CMS. The figure shows that at comparable mean PAPs, the haemoglobin levels in South Americans is higher than in Han Chinese.
Proposal for a new terminology of diseases associated with high-altitude pulmonary hypertension
Footnotes
-
↵Previous articles in this Series: No. 1: Humbert M, Trembath RC. Genetics of pulmonary hypertension: from bench to bedside. Eur Respir J 2002; 20: 741–749. No. 2: Galiè N, Manes A, Branzi A. The new clinical trials on pharmacological treatment in pulmonary arterial hypertension. Eur Respir J 2002: 20: 1037–1049. No. 3: Chemla D, Castelain V, Hervé P, Lecarpentier Y, Brimioulle S. Haemodynamic evaluation of pulmonary hypertension. Eur Respir J 2002; 20: 1314–1331. No. 4: Eddahibi S, Morrell N, d'Ortho M‐P, Naeije R, Adnot S. Pathobiology of pulmonary arterial hypertension. Eur Respir J 2002; 20: 1559–1572. No. 5: Widlitz A, Barst RJ. Pulmonary arterial hypertension in children. Eur Respir J 2003; 21: 155–176. No. 6: Moloney ED, Evans TW. Pathophysiology and pharmacological treatment of pulmonary hypertension in acute respiratory distress syndrome. Eur Respir J 2003; 21: 720–727. No. 7: Barberà JA, Peinado VI, Santos S. Pulmonary hypertension in chronic obstructive pulmonary disease. Eur Respir J 2003; 21: 892–905. No. 8: Dorfmüller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial hypertension. Eur Respir J 2003; 22: 358–363.
- Received May 12, 2003.
- Accepted May 30, 2003.
- © ERS Journals Ltd
References









