





Abstract
Prescribing corticosteroids for asthma based on mechanism-based endotypes rather than trait-based phenotypes http://ow.ly/HiHq30cA6Da
It is well known that corticosteroids improve asthma in a subgroup of patients and that this responder subgroup is invariably large enough to drive statistically significant differences in asthma outcomes when steroids are compared to placebo in unselected patients. The consistency of clinical trial data showing benefit from corticosteroids, especially in less severe patients, is the reason why asthma treatment guidelines consistently advocate their daily use in all but the mildest forms of asthma. Guidelines also typically advocate an empiric approach to corticosteroid dosing in which the dose of corticosteroid is increased as disease severity worsens. Such empiric approaches to treatment are now considered flawed and the emphasis is switching to development of treatment approaches that are guided by mechanism-based molecular endotypes rather than trait-based clinical phenotypes. The reason is simple: asthma traits such as symptoms and airflow limitation result from heterogeneous molecular mechanisms, and a one-size-fits all treatment paradigm to treat these traits does not make sense.
As the emphasis in treatment turns toward precision, the search for biomarkers that predict corticosteroid responders is intensifying. This search is guided by knowledge of the mechanisms by which corticosteroids improve asthma. Chief among these are the effects of corticosteroids on various members of the type 2 inflammation pathway. For example, corticosteroids suppress type 2 cytokines and the multiple effects of these cytokines on airway epithelial cells, including increases in nitric oxide levels in exhaled breath [1]. Corticosteroids are also effective in promoting eosinophil apoptosis. These data provide the rationale for the Berthon et al. [2] study reported in the current issue of the European Respiratory Journal. These authors examined how well a six-gene expression biomarker signature in induced sputum predicts lung function responses to high dose oral prednisolone in 55 adults with stable asthma. Among the six genes, one was a marker of eosinophils (Charcot Leyden crystal) and one was a marker of mast cells (carboxypeptidase A3). They found that 45% of patients responded to prednisolone and that the responder subgroup was characterised by airway and systemic eosinophilia and lower lung function. In addition, the six-gene expression biomarker signature was better at predicting steroid response than eosinophil numbers in blood and sputum. These data support the concept that asthmatics with airway type 2 inflammation (“Th2-high asthma”) are more responsive to corticosteroids than those with Th2-low asthma. But the data also show that markers of airway type 2 inflammation don't always predict responsiveness to steroids. For example, 41% of the non-responders had eosinophilic asthma at baseline, and 17% of patients treated with prednisolone had high sputum eosinophils after treatment. These data are consistent with multiple reports of persistent eosinophilia in subgroups of asthmatics despite treatment with high doses of steroids. Thus, lack of response to corticosteroids in asthma occurs in two clinical scenarios: one is a patient who does not have a responsive endotype (Th2-low asthma), and the other is a subset of patients who have Th2-high asthma.
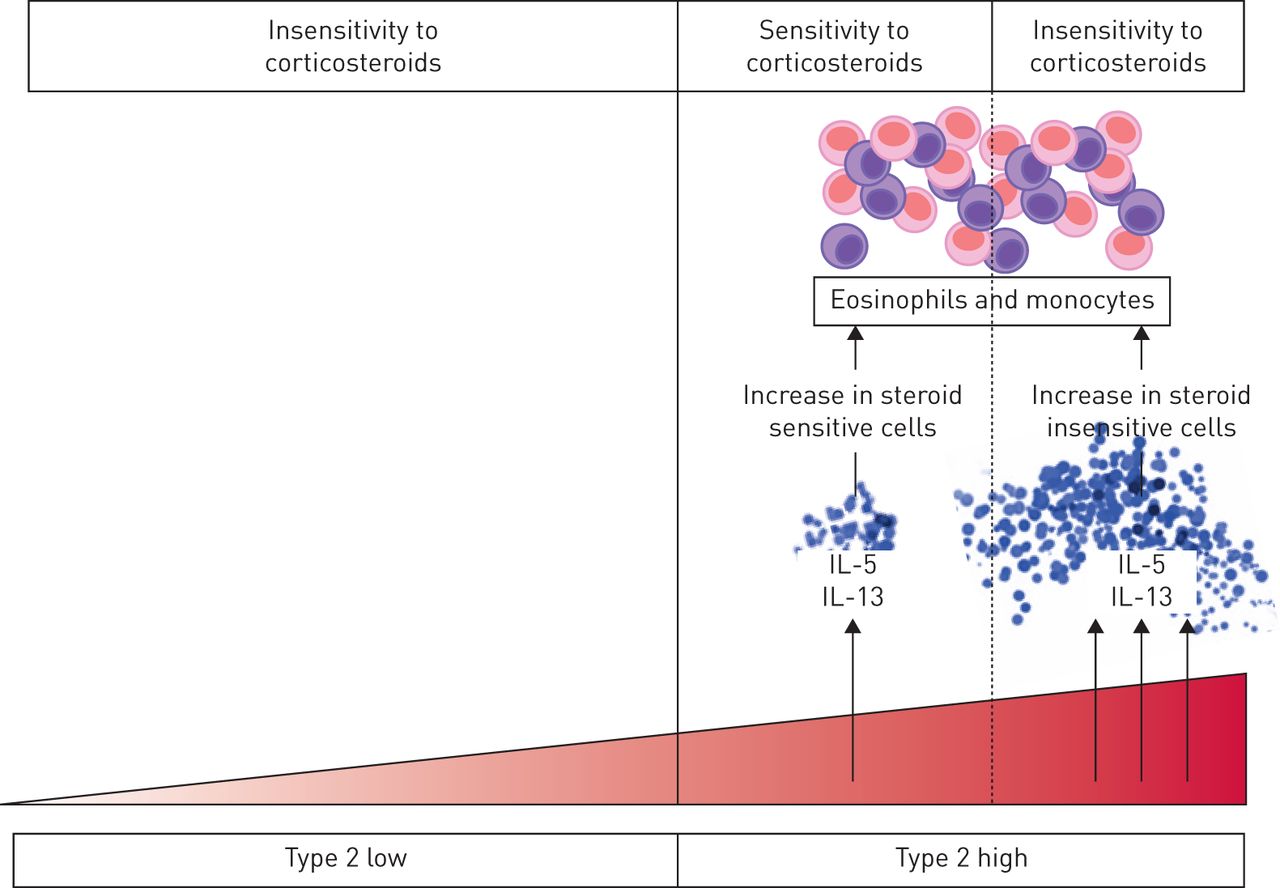
There are multiple reasons why a subset of Th2-high asthma patients may not respond well to corticosteroids, including lack of medication adherence. But it would be unwise to assume that poor adherence is the major reason for this phenomenon, because it would ignore possible biological reasons that could underpin new treatment approaches. One biological mechanism is that type 2 cytokines promote corticosteroid resistance. For example, although corticosteroids cause eosinophil apoptosis, this apoptosis effect is delayed in vitro by low dose IL-5 stimulation and is completely inhibited in eosinophils exposed to high dose IL-5 [3]. Thus, in vivo in asthma there may be a threshold effect for IL-5 above which it has biological effects that blunt steroid efficacy, and these biological mechanisms may not be overcome by higher dose steroid treatment (figure 1). In this scenario, targeting IL-5 becomes more effective than increasing steroid dose, and this may help explain the known clinical efficacy of IL-5 inhibitors in asthma. This example for IL-5 is the best described mechanism for type 2 cytokine and steroid resistance, but there is similar data for IL-13 pretreatment of monocytes suppressing the effects of steroids on lipopolysaccharide-induced IL-6 [4].
{kind=link}
Schematic for how steroid resistance in asthma increases as type 2 inflammation increases. Steroids have limited efficacy in type 2 low asthma. Among type 2 high patients, there may be a threshold effect for IL-5 and IL-13 above which these cytokines have biological effects that render eosinophils and monocytes less sensitive to steroids. This may explain persistence of airway eosinophilia in a subgroup of type 2 high patients treated with high doses of steroids.
Although effective in a subgroup of patients with asthma, it is now clear that steroids are not a one size fits all option. Despite this realisation, steroids remain a mainstay of asthma treatment and are often prescribed (per guidelines) in higher and higher doses to patients who do not respond well to low doses. The data on hand, including that from Berthon et al. [2], do not support this approach. Rather, the data support a treatment paradigm in which the dose of steroids should not be increased in patients who do not show increases in biomarkers of type 2 inflammation. Absent abnormalities in these biomarkers, it is hard to justify empiric increases in steroid dose in patients with asthma and the side effects that accompany the higher doses. And even with positive biomarkers of type 2 inflammation, we should be open to considering earlier use of specific inhibitors of type 2 pathway members (IgE, IL-4, IL-5, IL-13, CRTH2), because inhibitors of type 2 cytokines, in particular, may promote steroid sensitivity and allow patients to be treated with lower and less harmful doses of steroids. At present, the cost of these specific type 2 inhibitors may prohibit this approach on economic grounds, but it will hopefully become more feasible as the cost declines over time and as the marketplace becomes more competitive.
Disclosures
Supplementary Material
John V. Fahy ERJ-01167-2017_Fahy
Footnotes
Conflict of interest: Disclosures can be found alongside this article at erj.ersjournals.com
- Received June 12, 2017.
- Accepted June 12, 2017.
- Copyright ©ERS 2017










