Article Text

Abstract
Objectives Familial chilblain lupus is a monogenic form of cutaneous lupus erythematosus caused by loss-of-function mutations in the nucleases TREX1 or SAMHD1. In a family without TREX1 or SAMHD1 mutation, we sought to determine the causative gene and the underlying disease pathology.
Methods Exome sequencing was used for disease gene identification. Structural analysis was performed by homology modelling and docking simulations. Type I interferon (IFN) activation was assessed in cells transfected with STING cDNA using an IFN-β reporter and Western blotting. IFN signatures in patient blood in response to tofacitinib treatment were measured by RT-PCR of IFN-stimulated genes.
Results In a multigenerational family with five members affected with chilblain lupus, we identified a heterozygous mutation of STING, a signalling molecule in the cytosolic DNA sensing pathway. Structural and functional analyses indicate that mutant STING enhances homodimerisation in the absence of its ligand cGAMP resulting in constitutive type I IFN activation. Treatment of two affected family members with the Janus kinase (JAK) inhibitor tofacitinib led to a marked suppression of the IFN signature.
Conclusions A heterozygous gain-of-function mutation in STING can cause familial chilblain lupus. These findings expand the genetic spectrum of type I IFN-dependent disorders and suggest that JAK inhibition may be of therapeutic value.
- Autoimmunity
- Inflammation
- Cytokines
- Systemic Lupus Erythematosus
Statistics from Altmetric.com
Introduction
Familial chilblain lupus is a rare genodermatosis characterised by cold-induced erythematous skin lesions in acral locations.1 Histological findings include perivascular inflammatory infiltrates with deposits of immunoglobulins or complement consistent with cutaneous lupus erythematosus.1 Patients may develop arthralgia, antinuclear antibodies or lymphopenia. In contrast to sporadic forms, familial chilblain lupus manifests in early childhood and is inherited as autosomal dominant trait. Causative genes implicated so far include TREX12–4 and SAMHD15 which encode intracellular nucleases. TREX1 or SAMHD1 deficiency causes activation of antiviral type I interferon (IFN) induced by immune recognition of unmetabolised nucleic acids.6–9
Recently, activating de novo mutations in STING encoding stimulator of IFN genes, a signalling molecule of the cyclic GMP-AMP synthase (cGAS)-dependent DNA sensing pathway, have been reported in an autoinflammatory syndrome referred to as STING-associated vasculopathy, infantile-onset (SAVI).10–12 Patients presented with necrotising acral vasculitis, fever episodes and interstitial lung disease.12 Furthermore, a family with autoinflammation and lupus-like features segregating an activating STING mutation was reported,13 highlighting the role of the type I IFN system in the pathogenesis of innate and adaptive immune dysregulation.
Here we report a heterozygous STING mutation in familial chilblain lupus and demonstrate that it acts as gain-of-function mutation causing constitutive type I IFN activation. Moreover, we demonstrate that treatment of patients with the Janus kinase (JAK) inhibitor tofacitinib suppresses systemic type I IFN activation.
Materials and methods
For more details, see the online supplementary methods.
supplementary data
Study participants
Written informed consent was obtained by all participating members of the family or their parents. Patients were enrolled in an ongoing study on the pathogenesis of chilblain lupus approved by the ethics committee of the Medical Faculty, Technische Universität Dresden.
Statistical analysis
Statistical significance was determined by Student’s t-test. Values of p<0.05 were considered statistically significant. Data are represented as means±SEM.
Results
Clinical and laboratory findings
We describe a four-generation Greek kindred with five family members affected with chilblain lupus. Patients presented with cold-induced violaceous papules or plaques on fingers, toes, nose, cheeks and ears or with indurated purple patches on the thighs (table 1, figure 1A and see online supplementary figure S1). In some cases, recurrent ulceration led to necrotic destruction of distal phalanges of fingers or to mutilating lesions of the ear (figure 1A). Nailfold capillaroscopy showed irregular capillary loops with a tortuous appearance consistent with microvascular involvement (see online supplementary figure S1). Histological examination of skin from patients II.2 and III.2 revealed lymphohistiocytic cells within the perivascular region along with expression of the type I IFN-induced myxovirus resistance protein A particularly within the endothel (figure 1A). There were no haematological findings, but four out of five patients had low-titre antinuclear antibodies and one patient had high anti-C1q-antibodies (table 1). Fever episodes or internal organ manifestations were absent. In particular, none of the patients had a history of interstitial lung disease and pulmonary function tests including spirometry, body plethysmography and carbon monoxide diffusing capacity were unremarkable in all five affected family members. In addition, a CT of the chest performed in patient II.2 was normal.
Clinical findings of family members affected with chilblain lupus

Identification of a heterozygous STING mutation in familial chilblain lupus. (A) Inflammatory cutaneous lesions of individual II.1 on the nose, fingers and ear lobe with scarring deformities due to recurrent ischaemia and ulcerations. Histological analysis of skin (bottom) from individual III.2 shows an inflammatory perivascular infiltrate (H&E) as well as expression of the interferon-induced myxoma resistance protein 1 particularly within the endothelial layer of vessels. (B) Pedigree of the family with chilblain lupus. Solid symbols indicate affected, open symbols unaffected relatives, squares male persons, circles female persons and slashes deceased persons. Stars indicate genotyped individuals. Electropherograms (middle) depict sections of wild-type and mutant STING sequence with the heterozygous Gly166Glu mutation which fully segregates with chilblain lupus. Schematic of STING protein (bottom) with functional domains shows position of Gly166 within the dimerisation domain. (C) The modelled 3D structure of the human STING homodimer carrying the Gly166Glu mutation (green) displays an increased dimer interface (yellow surface) compared with the wild-type dimer (red). The mutated Glu166 residue of one monomer establishes hydrogen bonds with residues Thr263 and Thr267 (sticks) of the contralateral monomer that do not exist in the wild-type homodimer. CTT, C-terminal tail.
Identification of a heterozygous mutation of STING
After excluding TREX1 and SAMHD1 mutations in the family, we performed whole exome sequencing in seven family members (I.1, II.1, II.2, II.3, III.1, III.3, III.4) to determine the disease-causing mutation (figure 1B). Variant analysis and filtering assuming a dominant mode of inheritance revealed a heterozygous mutation in STING (c.497G>A; p.Gly166Glu, p.G166E) (figure 1B). This variant had previously not been annotated in the ENSEMBL or ExAC databases and was not present in 200 German control individuals. Sanger sequencing of this variant in other available family members revealed complete segregation with chilblain lupus. Multiple sequence alignment demonstrated that G166E affects a highly conserved residue within the dimerisation domain of STING14 (figure 1B and see online supplementary figure S2).
Structural analysis of mutant STING
We derived a structural model of the G166E STING monomer and its dimer interface by combining homology modelling with docking simulations using the crystal structure of human STING as template.15 Structural analysis revealed that substitution of glycine with glutamic acid at position 166 induces two pairs of hydrogen bonds between the Glu166 of each monomer and two threonine residues (Thr263 and Thr267) of the respective contralateral monomer (figure 1C and see online supplementary figure S3). Moreover, while the wild-type dimer interface is exclusively established by non-polar interactions,14 structural changes within the G166E homodimer give rise to additional 12 hydrogen bonds propagating from the mutated residue along the dimer interface (see online supplementary figure S3). Thus, G166E causes multiple previously non-existent intramolecular polar interactions augmenting the intermonomer contact surface area.
Mutant STING causes constitutive type I IFN activation
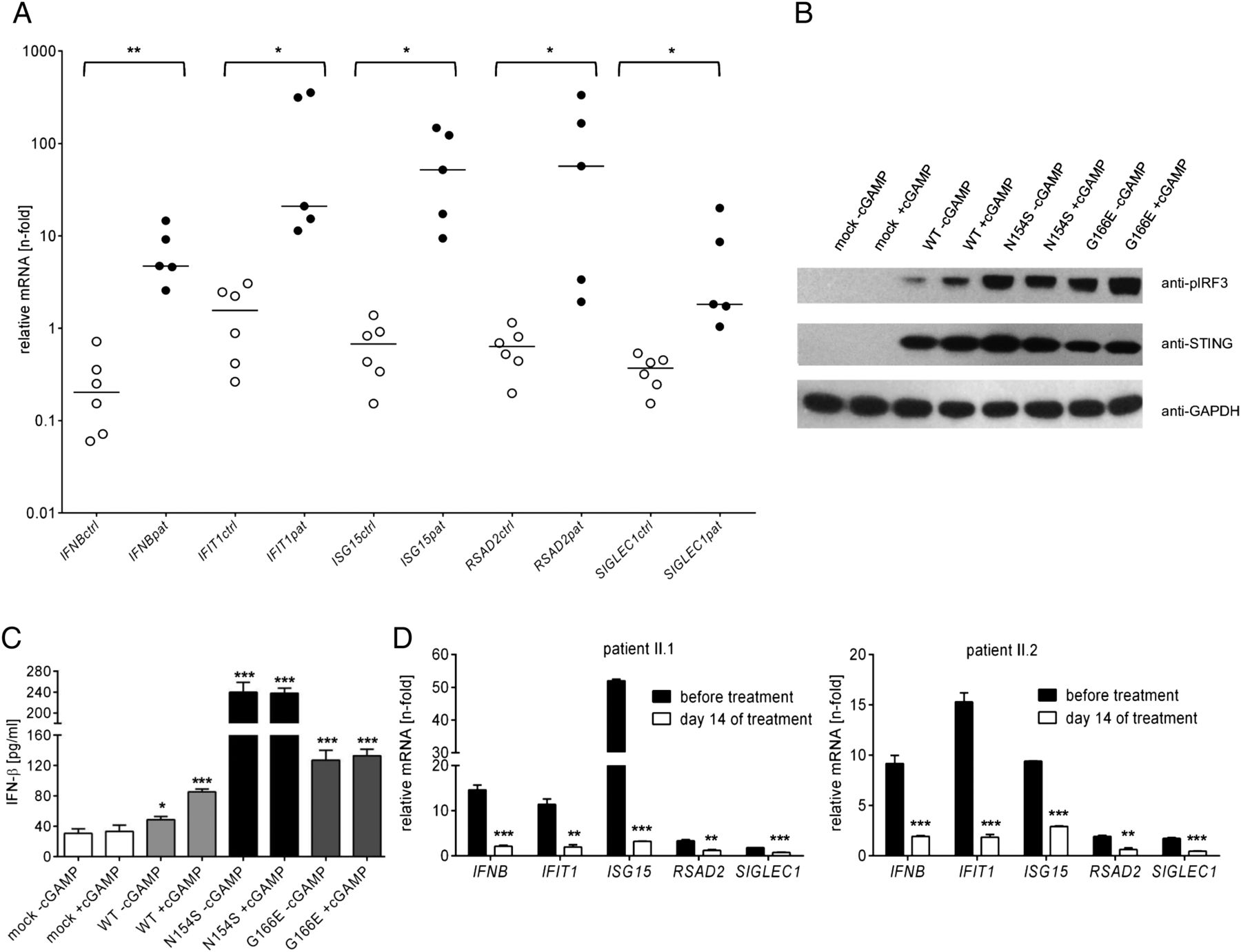
Binding of the second messenger cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) induces activation of the STING dimer and subsequent phosphorylation of interferon regulatory factor 3 (IRF3) through TANK-binding kinase 1 which results in transcriptional activation of the IFNB gene.12 ,16 We therefore investigated affected individuals for signs of an IFN signature in blood. Quantitative RT-PCR of peripheral blood cells revealed enhanced expression of the IFNB gene and of the IFN-stimulated genes, IFIT1, ISG15, RSAD2 and SIGLEC1 in all affected individuals examined, but not in unaffected family members (figure 2A and see online supplementary figure S4) demonstrating constitutive type I IFN activation.
{kind=link}
{kind=link}
Characterisation of functional consequences of the Gly166Glu STING mutation in vitro and in vivo. (A) Expression of the IFNB gene and the interferon (IFN)-stimulated genes, IFIT1, ISG15, RSAD2 and SIGLEC1, in peripheral blood of affected (pat, solid circles) compared with non-affected (ctrl, open circles) members of the family with chilblain lupus. Samples were run in triplicates. Horizontal lines indicate medians. *p<0.05; **p<0.01, Student’s t-test. (B and C) Effects of stimulation with the STING ligand cGAMP on type I IFN activation in HEK293T cells transfected with either empty vector (mock), wild-type STING (WT), the SAVI mutant N154S or the chilblain lupus mutant G166E. In response to cGAMP stimulation, cells expressing N154S and G166E exhibit increased phosphorylation of IRF3 (B) and enhanced levels of IFN-β (C) compared with cells expressing wild-type STING, both in the presence and absence of cGAMP. Western blots were also probed with antibodies against STING and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as loading control. Shown are the means and SEM of two independent experiments run in triplicates. *p<0.05; ***p<0.001 versus mock, Student’s t-test. (D) Effects of treatment of two affected individuals (II.1, II.2) with tofacitinib on the expression of IFN-stimulated genes in peripheral blood. Patients were treated with 5 mg tofacitinib per os twice daily. Samples were run in triplicates. Shown are the means and SEM. **p<0.01; ***p<0.001 versus before treatment, Student’s t-test.
To further explore the effects of G166E on the type I IFN axis, we transiently transfected HEK293T cells, which normally do not express STING, with human STING. On cGAMP stimulation, cells expressing wild-type STING responded with type I IFN activation as shown by increased IRF3 phosphorylation and IFN-β production (figure 2B, C). In contrast, cells expressing the SAVI-associated N154S mutation exhibited a strong baseline activation of IRF3 and IFN-β that did not further increase on cGAMP stimulation consistent with the previously reported activating nature of this mutation.12 Similarly, cells expressing the G166E mutation demonstrated a marked type I IFN activation in the absence of cGAMP (figure 2B, C), indicating that it acts as gain-of-function mutation.
Treatment with tofacitinib suppresses type I IFN signature
Binding of IFN-β to the IFN-α/β receptor, IFNAR, initiates the JAK/signal transducers and activators of transcription (STAT) pathway through activation of JAK1 and tyrosine kinase 2 (TYK2) which phosphorylate the IFNAR.16 ,17 This induces phosphorylation of STAT2 and STAT1 which in turn dimerise and translocate to the nucleus to activate transcription of IFN-stimulated genes. Given the chronic activation of type I IFN caused by the G166E mutation in the family with chilblain lupus, we assessed the therapeutic effect of the JAK inhibitor tofacitinib. With written informed consent, we treated two affected individuals with tofacitinib at a dose of 5 mg twice daily per os over 17 days. Assessment of IFN-stimulated genes in blood at day 14 revealed a strong suppression of the IFN signature (figure 2D) indicating effective systemic type I IFN inhibition. During the course of treatment, patients also reported a reduction of discomfort and pain of the fingers suggesting improved ischaemia.
Discussion
While familial chilblain lupus is a rare disease with only eight families carrying mutations in TREX1,2–4 and one family with SAMHD1 mutation5 described so far, understanding the underlying molecular pathology has revealed novel pathways of the intracellular nucleic acid metabolism that negatively regulate the type I IFN axis thereby preventing autoinflammation and autoimmunity,18 a key event in the pathogenesis of common forms of lupus erythematosus as well. Here we describe a gain-of-function STING mutation in familial chilblain lupus. As STING constitutes a signalling molecule that relays danger signals initiated by recognition of cytosolic DNA to activate type I IFN,11 these findings extend the genetic causes of familial chilblain lupus to components of the type I IFN signalling pathway and establish familial chilblain lupus and SAVI as different facets of a common disease spectrum.
All STING mutations described to date are localised within the dimerisation domain and are associated with fever episodes and interstitial pulmonary disease, in addition to signs of peripheral vasculitis.12 ,13 ,19 In contrast, symptoms of patients with familial chilblain lupus carrying the G166E mutation are confined to the skin, although systemic involvement is evident from an increased expression of IFN-stimulated genes in blood and formation of antinuclear antibodies. As shown by functional analysis, G166E acts as a gain-of-function mutation, like SAVI-associated STING mutations. Thus, G166E induces new intramolecular polar interactions that enhance dimer stabilisation resulting in constitutive type I IFN signalling in the absence of cGAMP ligand. However, the degree of type I IFN activation exerted by G166E is lower compared with the SAVI-mutation N154S suggesting a correlation between disease severity and the extent of inappropriate type I IFN activation.
Type I IFN receptor signalling is mediated by JAK1 and TYK2 which are the targets of JAK inhibitors,17 a novel class of immunomodulatory compounds such as ruxolitinib and tofacitinib approved for myelodysplastic disorders and rheumatoid arthritis, respectively.20 ,21 Interestingly, remission of symptoms of dermatomyositis and sporadic chilblain lupus was recently reported in two patients treated with ruxolitinib because of myelodysplasia.22 ,23 Moreover, both JAK inhibitors were shown to efficiently suppress type I IFN signalling in cells from patients with SAVI in vitro.12 Here we demonstrate for the first time that treatment with tofacitinib can effectively suppress type I IFN activation in patients with a genetically defined form of chilblain lupus caused by inappropriate type I IFN activation. Further clinical studies are required to assess the long-term benefits of JAK inhibitors for patients with inflammatory disorders triggered by type I IFN.
In summary, we identify a gain-of-function mutation in STING as a novel cause of familial chilblain lupus and demonstrate that constitutive type I IFN activation is amenable to treatment with tofacitinib. Our findings suggest that JAK inhibition may be of therapeutic benefit in other disorders with chronic type I IFN activation such as systemic lupus erythematosus.
Acknowledgments
The authors thank the patients and their family members for participation in the study. The authors thank Hanns-Martin Lorenz, Department of Internal Medicine, University of Heidelberg, for helpful discussion. The authors thank Peggy Binkenstein and Nick Zimmermann for excellent technical assistance. The authors thank Andreas Dahl and Mathias Lesche for next-generation sequencing and bioinformatics support.
References
Footnotes
Handling editor Tore K Kvien
NK and CF contributed equally.
Correction notice This article has been corrected since it was published Online First. The segmentation of the fifth author's name has been corrected.
Contributors NK, CF and MAL-K designed the study. NK, CF, CW, MS, KE and CG performed experiments and analysed data. ECC, VT, HAA and OC performed structural analysis. RG-M provided material and advice. NK and MAL-K wrote the paper. MAL-K supervised the study.
Funding Supported by grants from the Deutsche Forschungsgemeinschaft (LE 1074/3-1 and LE 1074/4-1, to MAL-K; TU 421/1-2 to VT, GU1212/1-1 and GU 1212/1-2 to CG), the Friede Springer Stiftung and the TU Dresden Graduate Academy (great!ipid4all) to MAL-K. OC is a career researcher of the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) Argentina. The Deep Sequencing Facility of the Biotechnology Center and the Center for Regenerative Therapies Dresden, Technische Universität Dresden, are funded by the Deutsche Forschungsgemeinschaft.
Competing interests None declared.
Ethics approval TU Dresden.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement NK and MAL-K have access to all data and data are available on request.