Article Text

Abstract
Objective: To determine the prevalence of systemic sclerosis associated pulmonary arterial hypertension (SScPAH), evaluate outcome, and identify predictors of mortality in a large patient cohort.
Methods: A prospective four year follow up study of 794 patients (722 from our own unit and 72 referrals). All patients screened for PAH using a combination of echocardiography, lung function testing, and clinical assessment. Patients with suspected raised pulmonary artery systolic pressures of >35 mm Hg, carbon monoxide transfer factor (Tlco) <50% predicted, or a precipitous fall in Tlco >20% over a one year period with no pulmonary fibrosis, and patients with SSc with breathlessness with no pulmonary fibrosis found were investigated with right heart catheterisation. All patients with SScPAH were treated in accordance with current best practice.
Results: The prevalence of PAH was 12% (89/722) by right heart catheter. The survival was 81%, 63%, and 56% at 1, 2, and 3 years from the diagnosis (in 89 patients from our own cohort and 59/72 referrals). Haemodynamic indices of right ventricular failure—raised mRAP (hazard ratio 21), raised mPAP (hazard ratio 20), and low CI (hazard ratio 11) predicted an adverse outcome There was no significant difference in survival between patients with SScPAH with (n=40) and without (n=108) pulmonary fibrosis (p=0.3).
Conclusions: The prevalence of SScPAH in this cohort was similar to that of other catheter based studies and lower than that of previous echo based studies. The 148 patients with SScPAH actively treated had comparable outcomes to those of the cohorts with primary pulmonary hypertension. A high mRAP was the strongest haemodynamic predictor of mortality. To improve prognosis, future treatments need to be implemented at an earlier disease stage to prevent right ventricular decompensation.
- systemic sclerosis
- pulmonary arterial hypertension
- survival
- iloprost
- CCB, calcium channel blocker
- HRCT, high resolution computed tomography
- PAH, pulmonary arterial hypertension
- PAP, pulmonary artery pressure
- PASP, pulmonary artery systolic pressures, PFTs, pulmonary function tests
- PPH, primary pulmonary hypertension
- PVR, pulmonary artery vascular resistance
- SSC, systemic sclerosis
- Tlco-carbon monoxide transfer factor
Statistics from Altmetric.com
- CCB, calcium channel blocker
- HRCT, high resolution computed tomography
- PAH, pulmonary arterial hypertension
- PAP, pulmonary artery pressure
- PASP, pulmonary artery systolic pressures, PFTs, pulmonary function tests
- PPH, primary pulmonary hypertension
- PVR, pulmonary artery vascular resistance
- SSC, systemic sclerosis
- Tlco-carbon monoxide transfer factor
Until recently, pulmonary arterial hypertension (PAH) in association with a connective tissue disease such as scleroderma (SSc) was regarded as a virtually untreatable condition.1 Fortunately, effective treatments are now becoming available. Badesch recently published a randomised, open label trial of 111 patients with SSc treated either with conventional treatment alone or epoprostanol and conventional treatment for 12 weeks demonstrating improvements in haemodynamics, exercise capacity, and symptoms in the epoprostanol treated cohort.2 In the Breathe-1 trial the magnitude of efficacy of Bosentan was similar in the subpopulation with SSc associated PAH to that in those with primary pulmonary hypertension (PPH).3 Similarly, trials involving inhaled4 and subcutaneous5 prostacyclin analogues showed significant functional benefit in patients with SSc. One limitation of these intervention studies has been the scarcity of information on certain aspects of the natural history of SScPAH, particularly those regarding the immediate and long term prognosis of the disease. Previous studies suggested that SScPAH is more lethal than PPH, but those were hindered by inadequate population size and were conducted before epoprostanol was available. Stupi et al found a two year mortality of 60% in 20 patients with SScPAH compared with <10% in a background population6 and Koh et al found a median survival of one year in 17 patients with SScPAH.7 Denton and MacGregor et al have suggested from echocardiographically based studies that the prognosis may be poor.8,9 Small, uncontrolled studies on mixed groups of patients with SSc, systemic lupus erythematosus and mixed connective tissue disorder10–12 support this conclusion. This lack of a large population size leading to loss of prognostic power has been addressed in PPH by the development of a registry format supervised by the US National Institute of Health.13–15 This approach has allowed a reliable determination of outcome in PPH and has led to the identification of haemodynamic variables that determine prognosis in an individual patient with PPH—namely, cardiac index, right atrial pressure, and pulmonary artery pressure. Hitherto in SScPAH, the link between haemodynamic parameters and prognosis has not been demonstrated.
Remarkably little is known of the natural history of SScPAH owing to a lack of longitudinal studies involving sufficient numbers of patients over a substantial time period. Previous prevalence rates have reported a wide variation of between 7 and 50%. Some investigators have used echo and others cardiac catheterisation as their chosen method of detection. Most have excluded patients with pulmonary fibrosis. Using echo as their chosen diagnostic method, Murata et al and Battle et al have found high prevalence rates of 35–49% in a total of 55 patients with SSc.16,17 Prevalence rates have been consistently lower when cardiac catheterisation has been used as the diagnostic tool. Murata’s prevalence rate dropped from 43% by echo to 29% when the same patients were catheterised (8/28). Similarly, Ungerer et al found a prevalence rate of 16% (8/49) by cardiac catheter, using a mean pulmonary artery pressure (mPAP) cut off ⩾22 mm Hg to define PAH.18 Salerni et al catheterised 10 clinically suspected patients and indicated a prevalence of 7%.19 An accurate estimation of prevalence is important for service planning purposes given the significant financial impact of new treatments. Our study aimed at (a) documenting the prevalence of PAH in a large cohort of patients with SSc; (b) determining the survival of patients with SScPAH as measured by pulmonary artery pressure on cardiac catheterisation; and (c) identifying the haemodynamic predictors of mortality in this population.
METHODS
Setting
Patients with SSc attending the Royal Free Hospital Connective Tissue Disease Clinic, London, were studied between March 1998 and September 2002. The patients attending the unit included those who were regularly followed up at the unit as part of the management of their SSc. Additionally, patients with SSc were referred to the hospital from throughout the UK for assessment when it was suspected that they were developing PAH.
Patient selection and evaluation
Between March 1998 and September 2002, a total of 722 patients underwent serial echocardiography, lung function testing, and clinical assessment at this institution (table 1). Each patient fulfilled the American College of Rheumatology preliminary criteria for the diagnosis of SSc.20 Echocardiography and pulmonary function tests (PFTs) were regularly performed on this cohort as part of their routine screening. Pulmonary artery systolic pressures (PASP) of >35 mm Hg on echo were used as a screening cut off point. Additional clinical criteria were (a) a carbon monoxide transfer factor (Tlco) of 50% of predicted or a precipitous fall in the percentage predicted Tlco by 20% in the previous year with no pulmonary fibrosis and (b) unexplained dyspnoea.
Patient selection for determination of prevalence in a cohort of patients with SSc receiving regular follow up at our institution
Seventy two additional patients were referred to the institution for further evaluation during this time period as they fulfilled a combination of the above criteria. Patients were identified as having pulmonary fibrosis if they had extensive (grade 3/4) changes visible on high resolution computed tomography of the thorax as reported by McDonald et al21 and/or reduced total lung capacity of <70% predicted. Thromboembolic disease was excluded by performing a V/Q scan or spiral contrast computed tomography. Patients identified by the above combination of non-invasive tests and clinical assessments were invited for right heart catheterisation.
Echocardiography protocol
M mode and cross sectional echocardiography was performed with the patient in the left lateral position using a Hewlett Packard (Andover, MA, USA) echocardiogram. Non-imaging continuous wave signals were recorded with a Doptek (Southampton, UK) 2.0 MHz transducer. Tricuspid regurgitant flow was identified in continuous wave mode at the apex. The peak instantaneous pressure drop from the right ventricle to the right atrium was calculated for the peak signal velocity from the tricuspid regurgitant signal by the simplified Bernoulli equation. No estimate of right atrial pressure was used to add to the Bernoulli equation value obtained from the TR jet.
Right heart catheterisation and acute vasodilator challenge
Informed consent was obtained before the procedure. Patients were given an infusion of iloprost only if they met the criteria for precapillary PAH by the modified NIH definition of a resting mean pulmonary artery pressure (PAP) of greater than 25 mm Hg or an exercise induced mean PAP of greater than 30 mm Hg, in association with a mean wedge pressure of less than 14 mm Hg.22 If the wedge pressure showed sustained elevation, then a postcapillary cause was suspected and the patient went on to have a left heart catheter and was excluded from the present study.
A 7 F, triple lumen, balloon tipped, flow directed pulmonary artery catheter (Baxter Edwards, Irvine, CA) was introduced through the right femoral vein using the Seldinger technique under strict aseptic conditions. Serial measurements of central venous pressure, pulmonary artery systolic/diastolic/mean pressures, pulmonary capillary wedge pressures, and cardiac output (measured by thermodilution) were taken. The baseline assessments were repeated at five minute intervals until pulmonary artery vascular resistance (PVR) varied by ±10%. Systemic and mixed venous oxygen saturations were measured at baseline and after the infusion of iloprost. Graded infusions of iloprost at 2, 6, and 12 ng/kg/min were started at five minute intervals, with complete haemodynamic assessment being made at each dosage. Systemic pressure was measured non-invasively throughout (Dinamap1800; Critikon, Tampa, FL). The study was stopped if the systolic blood pressure dropped to <90 mm Hg and in such cases intravenous fluids were given and the patient closely monitored until the blood pressure normalised. Pulmonary vascular resistance (PVR) was calculated as (mPAP−PCWP)/CO×79.92 in dyne.s/cm5(where PCWP=pulmonary capillary wedge pressure, CO=cardiac output).
Patient management
All patients found to have PAH on cardiac catheterisation were given warfarin (international normalised ratio 2–3). Between 1998 and 2000 “responders” were defined as patients who had a PVR reduction of >20% using the conventional definition of Rich et al.23 From 2000 onwards, in accordance with a change in practice, only patients with an absolute mPAP reduction >10 mm Hg were defined as “responders”. These responders with no/minimal lung fibrosis on high resolution computed tomography (HRCT) were treated with calcium channel blockers (CCBs)—nifedipine—up to 180 mg/day or diltiazem—up to 720 mg/day—as tolerated according to blood pressure and side effects. Those who had extensive lung fibrosis were not treated with CCBs. Responders who were unable to tolerate >120 mg nifedipine or 300 mg of diltiazem or who did not respond with a reduction of pulmonary pressure of >10% in three months were considered to have failed to respond to CCB treatment. These patients, as well as the other non-responders were invited to either join one of the trials of oral, inhaled, or subcutaneous preparations of prostacyclin analogues or if they had NYHA class III or IV dyspnoea, started to receive continuous ambulatory iloprost (given that they met the predefined criteria and had funding approval for this drug). Diuretics (frusemide or bumetanide and spironolactone) and digoxin were used if there was overt right ventricular failure. If there was evidence of right heart failure, Pao2 between 60 mm Hg and O2 saturation <90% at rest, then patients were given long term oxygen treatment at 1–4 l/min.
Follow up
All patients found to have PAH on cardiac catheterisation were followed up in our pulmonary hypertension clinic. If a patient died the mode of death was recorded either by direct contact with the attending clinician or from a witnessed account from family members. Survival times were measured as the time interval between the first cardiac catheterisation and the recorded date of death, or survival to 15 September 2002, which was taken to be the date of censor. No patient was lost to follow up.
Statistical analysis
Survival curves were derived using the Kaplan-Meier method from the SPSS statistical package. Haemodynamic data were recorded onto an Excel spreadsheet and comparisons between groups were made using the two tailed t test. A p value <0.05 was taken to be significant.
RESULTS
Prevalence determination
From the 722 patients with SSc actively followed up at our institution, 164 were selected by a combination of clinical findings and investigations (echo and/or PFTs) to be at high risk of having precapillary PAH. Table 1 shows the method by which these patients were selected for catheterisation from the background population. Twelve were not catheterised as the procedure was clinically not justified (reasons included advanced age and multiple comorbidity) and five patients refused. Of the 147 who did undergo cardiac catheterisation, 36 did not meet the criteria for PAH (mPAP <25 at rest and <30 mm Hg on benchfly exercise testing) and 22 had pulmonary hypertension due to other causes—3 had thromboembolic pulmonary hypertension and were referred for endarterectomy and 19 had post-capillary pulmonary hypertension (9 had poor LV function with PCWP >15 mm Hg, 10 had diastolic dysfunction). Hence, 89 patients were diagnosed as having precapillary PAH on cardiac catheterisation, giving a prevalence of 12% (89/722).
As stated earlier, 72 patients were referred to our institution for investigation for pulmonary arterial hypertension. All 72 underwent cardiac catheterisation, 59 were found to have PAH. As these patients with SScPAH did not belong to the cohort receiving regular follow up at our institution, they were excluded from the determination of prevalence. However, as these 59 patients from other institutions and 89 patients from our own institution had PAH proved by catheterisation, this total of 148 patients was included in the survival analysis.
Survival in patients with SScPAH
The survival of all patients with SScPAH diagnosed between 1998 and 2002 was determined. Eighty nine patients were selected from the cohort receiving regular follow up at the institution, and 59 selected from the patients referred from other institutions. Table 2 shows the demographic and baseline haemodynamic characteristics of these 148 patients with SScPAH. Their mean (SEM) age was 66 (7) years with a 4:1 preponderance of women:men. Most of the patients were anticentromere antibody positive (ACA+) and had moderate PAH—the mRAP was 7.7 (SD 4.8) mm Hg, mPAP was 39.5 (SD 13.5) mm Hg, mean (SD) PVR was 687 (564) dyne.s/cm5, and mean cardiac index (CI) was 2.6 (1.4) l/min/m2. The overall one year survival was 81%, 2 year 63%, and 3 year was 56%. PAH was a late complication of SSc, with PAH being diagnosed 14 (5) years from disease onset (fig 1). On dividing the patients into tertiles of mPAP for the purpose of analysis (fig 2, table 3), rising PAP was related to increased mortality. Patients with an mPAP <32 mm Hg had 30% higher one and two year survivals than patients with mPAP >45 mm Hg. There was a significant difference between each group (p<0.01).
Determination of survival in SScPAH: baseline demographic characteristics of 148 patients with SScPAH (89 from our own institution and 59 referred from other institutions) on cardiac catheterisation
Survival according to three bands of mean pulmonary artery pressure (mPAP in mm Hg) in SSc cohort

Cumulative survivals of 148 patients with SScPAH between March 1998 and September 2002.

Kaplan-Meier survival curves for 148 patients with SScPAH divided into tertiles—47 with mPAP <32 mm Hg, 48 with mPAP = 32–44 mm Hg, and 53 with mPAP >45 mm Hg. There was a significant difference in mortality between the three groups (p<0.01 between each group).
The cohort consisted of patients with SSc whose disease was both pulmonary fibrosis related (n=40) and non-fibrosis related (n=108). As stated earlier, the distinction between those with fibrosis and those without/minimal fibrosis was made on HRCT grading. For patients with no lung fibrosis, the one year survival was 82%, two year was 67%, and three year was 59%. For patients with lung fibrosis and PAH, one year survival was 75%, two year was 56%, and three year was 50%. Although survival appeared to be diminished in fibrotic patients compared with non-fibrotic patients, this difference was not significant (p=0.3)(fig 3). Additionally, there were no significant differences in mPAP, mRAP, and CI between fibrotic and non-fibrotic patients.

Kaplan-Meier survival curves for 108 patients with isolated SScPAH and 40 patients with pulmonary fibrosis related SScPAH (p=0.3).
Predictors of mortality
Forty seven of the 58 deaths in our cohort were directly attributed to right ventricular failure. Non-survivors had significantly worse indices of right ventricular dysfunction (p⩽0.001) than survivors. Of the three haemodynamic indices (mRAP, mPAP, and CI), the step up Cox multivariate regression analysis showed that right atrial pressure was the strongest independent predictor of mortality. As there was a high correlation between mRAP and mPAP and between mRAP and CI, the latter two indices could not be separated as independent predictors. The hazard ratio for mRAP was 20.7, with a significance of p=0.0001.
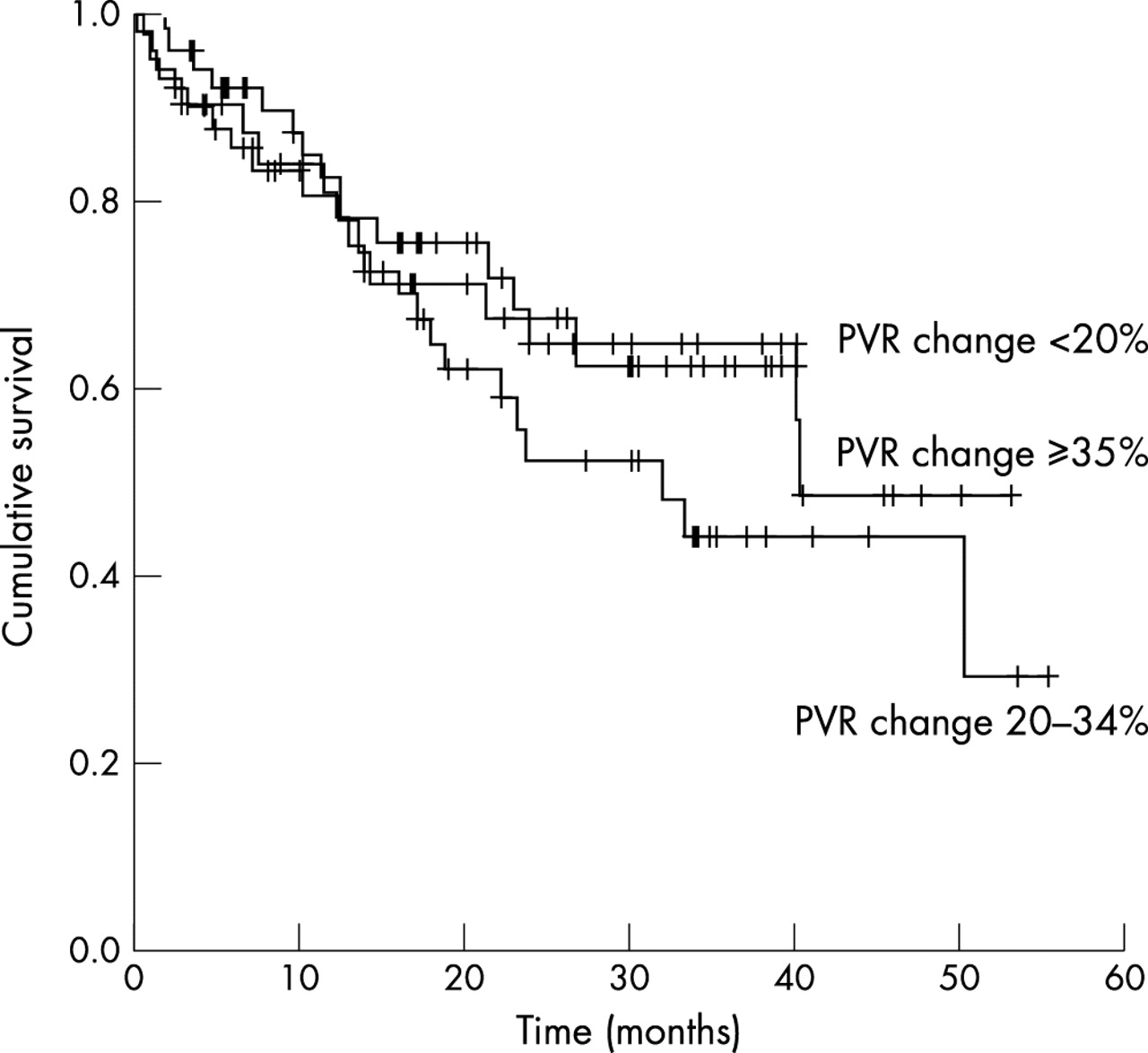
Vasodilator reserve to acute graded iloprost challenge was analysed using PVR falls of <20%, 20–34%, and ⩾35%. The division of patients into these three groups according to the degree of acute vasodilatation achieved on iloprost challenge was based on the findings of Raffy et al,24 who had previously demonstrated a correlation with vasoreactivity (indicated by the percentage fall in PVR) and survival in patients with PPH. In contrast with their findings, the degree of vasodilatation achieved after an acute iloprost challenge calculated by percentage fall in PVR (ΔPVR) did not predict survival in our SScPAH cohort. There was no significant difference between patients who had ⩾35% ΔPVR and those who had a <20% ΔPVR. (fig 4, table 4). Only 7 (5%) patients had a ΔPVR of >50%, and again their survival did not differ significantly from those with a ΔPVR <20%. Notably, doses of >120 mg/day nifedipine or 400 mg/day diltiazem were reached in 18 patients only because of side effects of gastro-oesophageal reflux, dizziness, flushing, and ankle oedema. The 40 patients who did not have >20% fall in PVR were managed as follows—22 received ambulatory iloprost at the onset and 18 were invited to participate in trials of oral, inhaled, and subcutaneous iloprost treatment, 4 patients received oral, 4 received inhaled and another 5 received subcutaneous iloprost, with 5 patients going on to receive ambulatory iloprost after initially receiving these trial drugs. The responders who were intolerant of CCBs were closely followed up with three monthly echo studies and if clinical/echocardiographic deterioration was suspected, they underwent repeat catheterisation with a view to prostacyclin treatment.
Survival according to percentage change in PVR (acute vasoreactivity) on acute iloprost challenge (see fig 4 and text)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Survival according to degree of vasoreactivity to acute iloprost challenge in 129 patients with SScPAH divided into tertiles of PVR change (ΔPVR)—40 patients had a ΔPVR of <20%, 50 had a ΔPVR of 20–34%, and 49 had a ΔPVR of ⩾35%. There was no significant difference in survival between the three groups (p=0.8).
DISCUSSION
As far as we know this is the largest cohort of patients with SSc comprehensively evaluated with serial echocardiography, lung function testing, and clinical evaluation followed up over a four year period. The prevalence of SScPAH by catheter criteria in our 722 patients was 12% (89/722). This finding concurs with that of Ungerer et al,18 who reported a prevalence of 16%. Their study was catheter based and used a background population of asymptomatic patients with SSc similar to our cohort. Given that our study was performed over a sufficiently long period of time with all patients accounted for during follow up, the prevalence rate of 12% is likely to be accurate. Echo based studies of Murata16 and Battle17 found prevalence rates of a higher magnitude. This might have been due to the limitations of their echo technique to estimate the tricuspid gradient, and the smaller population size. More recent observations suggest that echo determined tricuspid gradient correlated quite poorly with pulmonary pressures25 and increases in tricuspid gradient do not directly relate to mean pulmonary pressures.26 A further limitation of echo studies is that left heart disease with raised wedge pressures can contribute to apparent pulmonary hypertension, leading to overestimation of prevalence. Given this tendency towards overestimation of tricuspid gradient by echo studies, we felt that this investigation had a limited role in establishing the diagnosis but instead was a convenient non-invasive tool for identifying patients at high risk in combination with clinical assessment and declining Tlco.
We established outcome in a large group of 148 patients with SScPAH. These patients were selected from our own cohort of patients with SSc receiving regular follow up and also from patients referred to our institution for further investigations from other units. Follow up information was available on all patients. The cumulative survival in this population of patients with SScPAH treated with the currently best available treatments was 81% at one year, 63% at two years and 56% at three years. Not surprisingly, rising mPAP predicted outcome. Patients with an mPAP of >45 mm Hg at the time of the first catheterisation, had a 30% higher one year mortality than patients with mPAP <32 mm Hg.
We found that the haemodynamic parameters of right ventricular dysfunction at the time of catheterisation were strong predictors of outcome. This is the first study to identify haemodynamic parameters that predict prognosis in SScPAH. Our findings concur with those of D’Alonzo et al.14 who similarly identified mRAP, mPAP, and CI to accurately reflect prognosis in their cohort of patients with PPH gathered for the NIH registry. Identifying such predictors is important in clinical practice as it is helpful in identifying those patients who have a poor prognosis and require urgent transplant. In addition, it enables clinicians to give an accurate estimate of prognosis and determine future treatment pathways in individual patients with SScPAH.
The mortality found in our cohort was lower than that previously reported by Koh et al,7 who found a one year survival of 50% in 17 patients with SScPAH with a mean PAP of 45 mm Hg. In our subgroup of patients with a mean PAP >54 mm Hg (the group with mPAP >45 mm Hg) the one year survival was 61%. The reason for this difference in survival may be related to the fact that this was an actively treated cohort of patients with SSc, given epoprostenoids when indicated. In patients with PPH clear evidence has emerged that prompt epoprostenoid treatment can have an impact on prognosis. McLaughlin et al27 have recently reported impressive results in 162 patients with PPH, with 1, 2, and 3 year observed survivals of 88%, 76%, and 63%, with an average mPAP of 61 mm Hg in their cohort. The smaller improvement in survival in patients with SScPAH treated similarly with advanced treatments may be due to the systemic nature of disease involvement in SSc with right ventricular decompensation occurring at lower pulmonary artery pressures compared with PPH. From our continuing SScPAH registry, the impact of early intervention with epoprostenoids or a combination of advanced treatments remains to be assessed in the future, especially as the management strategies of such patients has changed significantly over the 1998–2002 period.
Limitations
Our study had several limitations. Firstly, this was a descriptive study of patients with SSc who were treated according to their clinical characteristics, not a controlled trial. Owing to limitations in the methodology, only general conclusions can be reached. However, the study does provide reasonably accurate background information on prevalence and outcome, which has not been previously reported in this rare disease, because of the large numbers of patients collected by the registry approach. Secondly, patient numbers diminished, particularly after two years of follow up, and hence when estimating mortality at >2 years predictions may be inaccurate. Even so, we have provided evidence that SScPAH is a disease which, even when it presents in the mild stage (mPAP <32 mm Hg), has a significant rate of progression. Thirdly, with regards to assessing the impact of treatment, survival in individual treatment groups was not analysed as the numbers of patients in each group were too small to give adequate statistical power.
CONCLUSIONS
Our estimation of prevalence of PAH in 722 patients with SSc actively treated and followed up is lower than that of previously reported smaller studies, especially those where echocardiography was the mainstay of diagnosis. Haemodynamic indices of right ventricular decompensation, namely mRAP, mPAP, and CI, predict survival in SScPAH. The role of CCBs is limited in this patient group owing to the lack of tolerability of high doses and the small numbers of responders to acute vasodilator challenge. Initial data suggest that the prognosis in patients with SScPAH is unaffected by the presence or absence of pulmonary fibrosis. Hence, there may be a role for aggressive treatment in this subgroup of patients with SScPAH.