


Abstract
Thalidomide was synthesised and launched several decades ago as a drug against respiratory infections and was administered to pregnant women for relief of morning sickness. The drug was withdrawn when its teratogenic effects came to light. Thalidomide and its analogues suppressed cell proliferation and angiogenesis and controlled invasion and metastasis of tumours in pre-clinical studies. With the recognition of its immunomodulatory and anti-inflammatory, properties, thalidomide may have found a place in the treatment of many forms of cancer and autoimmune conditions. Herein the signalling pathways modulated by thalidomides via the mediation of vascular endothelial growth factor, phosphoinositide-kinase/protein kinase B and nuclear factor kappa B, and mammalian target of rapamycin, which integrates these signalling systems, are discussed. The mode of action of thalidomides and their strategic utility in therapy are evaluated in the context of potential clinical benefits. Notwithstanding the perceived benefits, the side-effects of thalidomides need to be taken into account; they do exert teratogenic effects in animal models, although being effective at lower doses, the drugs seem to show comparatively manageable and reduced toxicity. Combination therapy of thalidomides and modulators of signaling that they influence may further reduce the severity of the side-effects by delivering inhibitory effects at reduced drug dosages. Pre-clinical evaluations of this kind seem warranted.
Thalidomide was synthesised in 1954 and launched in 1967 as a drug to deal with respiratory infections and was administered to pregnant women for relief of morning sickness. The drug was withdrawn when its teratogenic effects came to light in reports from numerous countries. We now know that the teratogenic effects are due to the binding of thalidomide to the protein called cereblon, which is found in embryonic and adult tissues. Cereblon is required in normal morphogenesis. Suppression of cereblon produces malformation similar to that encountered with thalidomide, and addition of a form of cereblon that does not bind thalidomide reduced the teratogenic effects (1). Thalidomide has since evolved into a drug with immunomodulatory, anti-inflammatory, anti-angiogenesis and cell proliferation inhibitory properties with potential use in the treatment of many forms of cancer and autoimmune conditions. Thalidomide analogues have been synthesised to combat toxicity and improve efficacy (2, 3).
Thalidomide and its analogues lenalidomide and pomalidomide, referred to herein as thalidomides, represent a class of drugs which possess anti-inflammatory and immunomodulatory properties. These compounds have been used in the management of patients with multiple myeloma, and pancreatic, prostate and lung cancer, as well as in autoimmune conditions such as graft-versus-host disease, Waldenstrom's macroglobulinaemia, experimental autoimmune encephalomyelitis (EAE) and suggested to be of potential benefit in the treatment of multiple sclerosis (MS). Thalidomides possess not only growth-inhibitory and anti-angiogenic properties but they are also active modulators of the immune system by virtue of which their potential use in the treatment of human disease has been recognised and investigated vigorously despite the early justifiably adverse publicity that thalidomide received. The focus here is to review the molecular basis of these phenotypic effects generated by thalidomides, identify the signalling systems which they modify and moderate, and to identify beneficial adjuvant therapies that might enhance the efficacy of thalidomides.
The thalidomides are capable of controlling biological features that are highly relevant in the context of tumour development and secondary spread (Table I). Of considerable interest is the inhibition of angiogenesis and cell proliferation, and promotion of apoptosis. Tumours and stromal cells produce many chemokines which promote tumour growth and induce angiogenesis. The chemokines are also associated with the evasion of immune surveillance by tumours. Thalidomides suppress the production of many chemokines and therefore they may combat immunological dysfunction associated with tumours. The ability to restore endogenous immune function has indeed been attributed to the thalidomides. These functional capacities may have contributed to the recognition of the potential of thalidomides for deployment as anticancer agents. Given the association of angiogenesis with MS (4), the thalidomides have been viewed as a therapeutic option in MS and other autoimmune conditions. The objective herein is to emphasise possible ways and means to resist or reduce the teratogenic side-effects by analysing the signalling systems which they down regulate and to identify modulators of signalling that might function in synergy with the thalidomides.
Salient biological features influenced by thalidomides.
Control of Angiogenesis by Thalidomide and Its Analogues
The ability of these immunomodulatory drugs is of considerable interest in the backdrop of their effects on angiogenesis and cell proliferation and tumour growth. Some while ago, thalidomide was shown to inhibit vascular endothelial growth factor (VEGF)- and basic fibroblast growth factor (bFGF)-induced angiogenesis in corneal angiogenesis assays (5, 6). Lu et al. further showed that lenalidomide-mediated inhibition of VEGF resulted in suppression of phosphoinositide-3 kinase (P13K)/protein kinase B (AKT) signalling and inhibited the formation of angiogenesis-related effects on primary endothelial cells in vitro (7). In vivo, lenalidomide suppressed lung colonisation of B16-F10 tumours.
Medinger et al. studied the clinical outcome in patients with plasma cell myeloma treated with the thalidomides and their combination with the proteasome inhibitor bortezomib (8). They looked at several angiogenic markers and the levels were consistent with the notion that the response rate of patients was attributable to anti-angiogenic effects exerted by the thalidomides. The clinical response was heightened by bortezomib which promotes apoptosis by phosphorylating and cleaving B-cell lymphoma 2 (BCL2) (9).
Consistent with these findings, thalidomide and the analogues lenalidomide and pomalidomide have been reported to inhibit in vitro migratory behaviour of colorectal carcinoma cells. These drugs also suppressed the formation of metastatic nodules in vivo in murine hosts (10). Subsequently, Liu et al. found that genes involved in immune response as well as cell adhesion signalling were affected by treatment with thalidomide analogues (11). They emphasised the alterations in NOTCH/WNT signalling and the regulatory genes Kremen 2 which regulates Dickkopf 2 (DKK2)-mediated inhibition of WNT signalling and Deltex 4 (DTX4), which is down-regulated by NOTCH. Liu et al. seem to imply that their findings might indicate a mechanistic membrane-associated alteration occurring as a consequence of treatment. The WNT–Frizzled receptor complex can activate many signalling systems including the canonical β-catenin pathway. Indeed, NOTCH does affect the function of cell adhesion molecules cadherins and regulates intercellular adhesion. Nevertheless, the caveat has to be considered that NOTCH and WNT proteins can exert opposing or co-operative effects in different systems. But, overall, NOTCH would inevitably modulate cell motility or invasive behaviour, cell proliferation and apoptosis. Furthermore, one should recognize that these thalidomides alter VEGF expression and influence cell proliferation via NOTCH1/DELTA4 or the PI3K/AKT pathways (7, 12).
Nuclear factor -κB (NF-κB) is another pathway implicated in thalidomide action. Keifer et al. demonstrated this over a decade ago (13). Thalidomide inhibited I kappa B Kinase (IKK) and NF-κB target genes interneukin 8 (IL8), the IL signal transducer tumour necrosis factor receptor associated factor 1 (TRAF1), and cellular inhibitor of apoptosis protein 2 (IAP2). Indeed, both thalidomide and lenalidomide may block NF-κB activation. Liu et al. noted inhibition of NF-κB together with marked increase in BCL-2 associated X (BAX)/BCL2 ratio and that the effect on the apoptosis family genes was more marked than NF-κB inhibition (14). Finally, as discussed below there is the perceived promotion by thalidomides of crosstalk between pathways of angiogenic signalling by mammalian target of rapamycin (mTOR), and this appears to possess therapeutic possibilities.

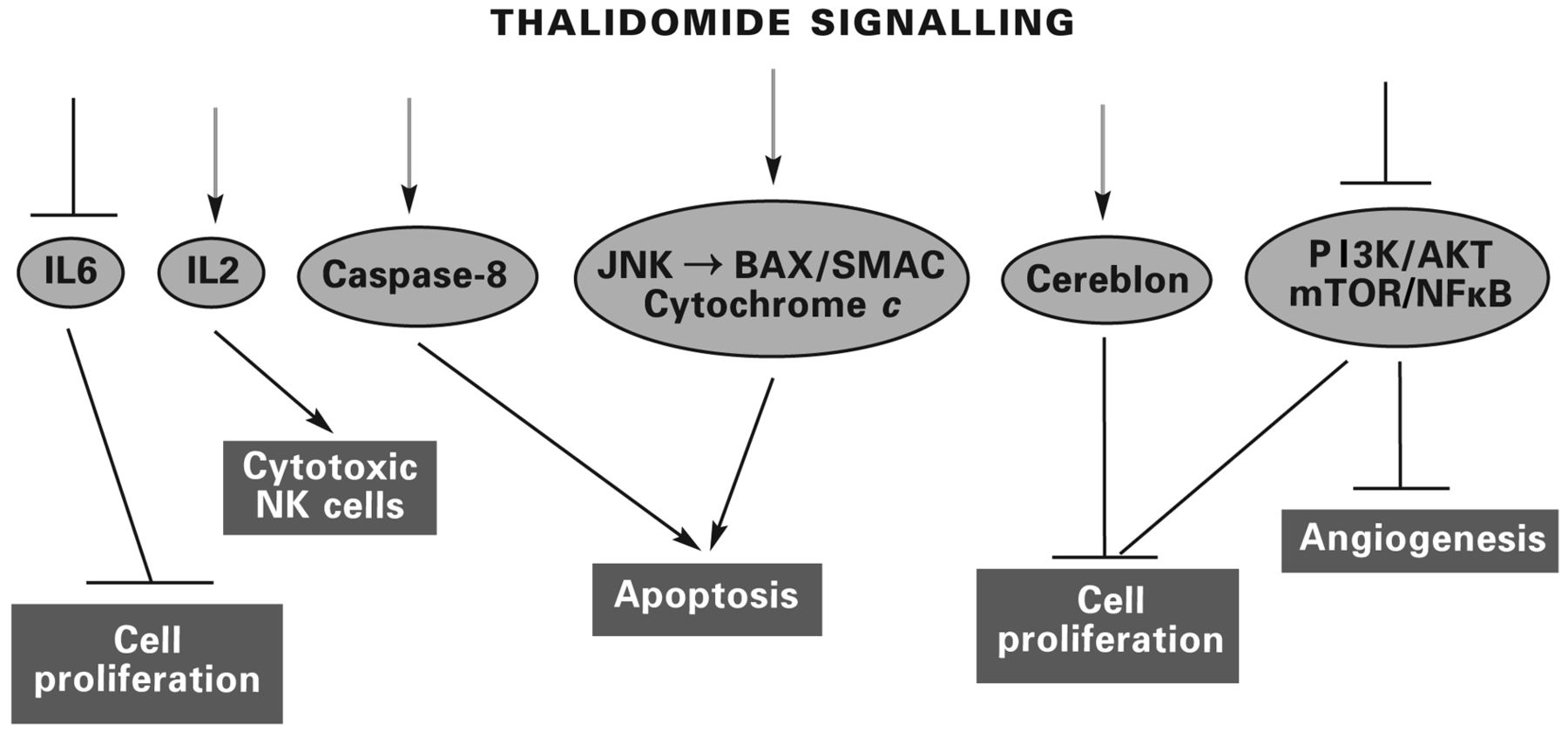
Pathways of thalidomide signalling in the control of cell proliferation and induction of apoptosis and inhibition of angiogenesis in myeloma and other tumour types. Other cytokines suppressed by thalidomide and the analogues include interleukin 8 (IL8) and tumour necrosis factor-α (TNFα). Some of the angiogenic pathways are integrated in their function by mammalian target of rapamycin (mTOR). SMAC is a mitochondrial protein that promotes the release of cytochrome c, leading to the activation of caspase-mediated apoptosis. Regulation of BCL-2 associated X (BAX) by Jun N-terminal kinase (JNK) is an essential element in this apoptotic signalling (18, 19 and references discussed in the text).
The mTOR Signalling Pathway Might Integrate with Thalidomide Function
The mTOR signalling pathway has been associated with several cellular processes, such as cell proliferation, growth, apoptosis, angiogenesis, cell motility and invasion (see Figure 1). It could facilitate cross talk between VEGF, PI3K/AKT and NF-κB, which are linked with angiogenesis. The integration of these signalling systems by mTOR has prompted assessment of the effects of mTOR inhibitors in combination with thalidomide and its derivatives. This integrative ability of mTOR would explain the synergy seen in the effects of everolimus and lenalidomide in a phase I clinical trial involving patients with refractory multiple myeloma. The combined therapy correlated with the down-regulation of mTOR in the responders with active mTOR signalling (15). Beneficial effects have been reported with the combination of temsirolimus with lenalidomide in another phase I trial involving patients with advanced forms of cancer, but this study relates to small numbers of patients with each tumour type (16). Median overall and progression-free survival of all evaluable patients were presented. No response was noted in those with head and neck squamous cell carcinoma, non-small cell lung carcinoma and endometrial carcinoma. The authors of that study concede the need to look at selected tumour types where stable disease was noted. They also state that abnormalities of PI3KCA, the catalytic subunit of PI3K, showed no correlation with benefits of therapy.
Suppression of Cell Proliferation and Tumour Growth by Thalidomides
That thalidomides can inhibit cell proliferation in vitro, in particular of endothelial cells, keratinocytes and some epithelial tumour cell lines, was shown some years ago, but the mechanisms involved in this inhibition were not elucidated until recently. Thalidomides seem to suppress cell proliferation by interference with cell-cycle regulatory components. Fecteau et al. showed that lenalidomide supressed chronic lymphocytic leukaemia (CLL) cells in vitro apparently by inducing the expression of p21 (17). The thalidomides are known to exert their effects by targeting the protein cereblon, whose role in the manifestation of their teratogenic effects was alluded to earlier. Cereblon is targeted by lenalidomide and when cereblon was silenced, the induction of p21 by lenalidomide was reduced. These findings suggest that the inhibition by lenalidomide of CLL proliferation occurred in a cereblon/p21-dependent manner. Cereblon is a component of the ubiquitin-E3 ligase Cullin 4–Ringbox protein 1-Damage specific DNA binding protein 1 (CUL4)–RBX1–DDB1) complex. It is directly targeted by thalidomides (1), leading to the recruitment of their target zinc finger transcription factors to the ubiquitin-E3 complex, their ubiquitination and degradation, consequently suppressing cell proliferation and cell viability.
Immunomodulation by Thalidomides
Cancer development and progression occur as a consequence of loss of surveillance attributable to a progressive suppression of the immune system. In contrast, pathogenesis of autoimmune conditions results from a loss of tolerance to self antigens. Many individual factors contribute to the maintenance of immunological tolerance in cancer by forming a complex network of immune suppression in the tumour microenvironment, spreading to the associated lymphangiogenic system (20, 21). Tumours are known to produce ILs, transforming growth factor (TGFβ), and VEGF, all able to suppress the function of the immune system. Notably, the differentiation and maturation of antigen-presenting dendritic cells are impaired, with resultant impairment of their interactions with T- and B-lymphocytes and natural killer (NK) cells (21, 22).
The strategy of immunotherapy for cancer has been evolving over many years. The administration of monoclonal antibodies, treatment with cytokines, and using immunomodulatory drugs to stimulate the immune system are among important therapeutic approaches. Much effort has gone into the development and deployment of immune modulators that can target immune checkpoints and re-activate endogenous immune responses. Immune resistance by tumours seems to arise from tumours being able to co-opt certain immune checkpoint pathways, especially against T-cells that are specific for tumour antigens. Immune checkpoint proteins regulate T-cell responses. The interaction between the receptor and ligand lead to the inhibition of T-cell activation and cytokine production. Therefore, blocking the function of the immune checkpoint proteins has been viewed as a viable therapeutic approach. The modulation of T-cell activation pathways has taken the form of using antibodies against the T-cell receptors or ligands (23). Ipilimumab an antibody raised against cytotoxic T-lymphocyte-associated antigen 4 (CTLA4) has received approval for therapeutic use. Other immune checkpoint proteins currently tested are programmed cell death protein 1 (PD1) and its ligand PDL1. Antibodies raised against PD1 and PDL2 are being clinically evaluated (24). Recently Gorgun et al. showed that lenalidomide accentuated the immune effects of PD1/PDL1 blockade in multiple myeloma (25). The heightened effects are mediated by cereblon, which is a component of one of the pathways discussed earlier in this review (Figure 1).
Besides being able to suppress angiogenesis and cell proliferation, the thalidomides appear to possess added advantage as immunomodulators and suppressors of inflammation in combating immune resistance by cancer, both haematological malignancies and solid tumours. Given that CD4+, CD8+ and γδ T-cells are important contributors to the cytokine scenario and that γδ T-cells possess anti-tumour properties, much effort has been devoted to induction of these T-cell subtypes in evolving therapeutic strategies against cancer and autoimmune diseases. However, the anti-tumour effects of γδ T-cells may be countermanded by IL17 and thus effectively turn them into tumour promoters (26).
De Keersmaecker et al. examined the response of CD4+ and CD8+ cells from patients with multiple myeloma to lenalidomide and pomalidomide (27). Both drugs increased proliferation of and cytokine production by CD4+ and CD8+, enhanced the lytic capacity of cytotoxic T-lymphocytes and reduced the suppressive effects of CD4+ regulatory T-cells on CD8+ responses.
Lenolidomide promoted proliferation of CD8+ cells, but then it also enhanced a population of myeloid cells which were CD1+/CD15+. These cells were capable of suppressing the proliferation of both CD8+ and CD4+ cells. This had led to postulate that lenalidomide might promote both activating and inhibitory components as a means of regulating the immune system (28). In the EAE model, T helper 1 (Th1) and Th17 CD4+ subtypes mediate pathogenesis but the CD8+ contribution is often underestimated (29). In the context of cancer, one should recall that target-specific CD8+ cells can be generated to inhibit angiogenesis. DNA vaccines using Salmonella to target VEGF receptor 2 of the tumour vasculature have been shown to inhibit tumour angiogenesis and metastasis (30). Jellbauer et al. induced CD8+ cells by immunising animals with recombinant Salmonella strain carrying a CD8+ epitope from VEGF receptor 2 (31). In the B16-F10 melanoma tumour model, the immunized animals showed induction of a VEGF receptor 2-specific CD8+ population which would have targeted the VEGFF receptor 2-expressing tumour vascular endothelium and reduced metastatic spread into the lungs and tumour growth. One can cite many instances of specific expression of growth factors or their receptors in tumours. Whether an indirect and somewhat involved approach to targeted destruction of tumour cells should be adopted would be based on the location of the tumour or the metastatic deposits and whether the target is accessible to therapeutic agents or specific antibodies.
Clinical Evaluation of Thalidomide and Derivatives
The clinical outcome in a lenalidomide trial of metastatic renal cell carcinoma conducted in 2004 was not very encouraging, with complete response in 3%, partial response in 8% and stable disease in 21% of the patients recruited. Secondary end-points of tumour progression, response duration and progression-free survival were unremarkable (32). Comparable with this was stable disease response in 57% obtained by Patel et al. (33). Vestermark et al. looked at response in patients with brain metastases of melanoma (34). No response was noticed in brain metastases and 3/35 patients showed partial response of extracraneal metastases. Combination of thalidomide and the derivatives with standard chemotherapy might be warranted. A few clinical trials are in progress designed to study the effects of thalidomide and its analogues on their own or in combination with other drugs in different neoplastic conditions. A phase I clinical trial is being carried out administering lenalidomide to patients with high-grade glioma. Combination treatment of CLL with lenalidomide and dexamethasome is also being clinically evaluated, as is combination with temsirolimus in patients with Hodgkin's and non-Hodgkin's lymphoma. The importance of thalidomides for the treatment of multiple myeloma and other B-cell malignancies cannot be over-emphasised. But treatment efficacy in solid tumours is uncertain. Response of some forms of tumour has been positive either with thalidomide on its own or in combination with conventional chemotherapy. The responses can vary among tumour types, with some not showing any response at all.
Notwithstanding the perceived potential benefits, the side-effects of therapy of thalidomide and derivatives need to be emphasised. They do exert teratogenic effects in animal models, although being effective at lower doses the analogues lead to comparatively manageable and reduced toxicity. With the objective of reduction of toxicity in mind, the designing of new analogues would need to consider an important aspect of pharmacology and that is that enantiomers of chiral drugs differ in respect to their pharmacokinetics and toxicity (35). Thalidomide is a chiral drug and its R and S enantiomers differ in their pharmacological effect. The R isomer is not believed to be teratogenic but the S enantiomer is. However, racemisation of the drug in vivo limits its use. Lee et al. have described a non-racemising derivative of thalidomide, fluorothalidomide, which is said to exert no teratogenic effects (36). However, it should be here recalled that previously the fluoro-derivative was reported to be toxic and to have no TNFα-inhibitory activity, whilst another derivative, α-fluoro-4-aminothalidomide, also chirally stable, was not cytotoxic but did markedly inhibit TNFα (37). Much needs to be done and much can be done in the domain of thalidomides to enhance their efficacy and reduce toxicity.
Concluding Thoughts
Thalidomide has an unenviable history of opprobrium and disrepute owing to its teratogenic effects. However, of late, the efficacy in suppressing invasion, angiogenesis and metastatic spread of cancer may have earned thalidomide and its analogues a place as potential therapeutic agents coupled with the strategic benefits of immunotherapy in the control of cancer. From the elucidation of signalling systems which are involved in their biological function has arisen the possibility of deploying thalidomide therapy in combination with modulators of those signalling pathways. It would be needless reiteration that the perceived potential benefits have to be balanced with the side-effects of therapy. The thalidomides do seem to exert teratogenic effects in animal models, but being effective at lower doses, the side-effects may be manageable and toxicity reduced. Combination therapy of thalidomides and modulators of the signalling systems they influence may further reduce the severity of the side-effects. Pre-clinical evaluations of this kind of therapy seem warranted.
Acknowledgements
I thank Dr M.S. Lakshmi for reading the manuscript and making helpful suggestions, and Professors Satnam Dlay and Barrie Mecrow for much help and support.
- Received July 6, 2015.
- Revision received August 11, 2015.
- Accepted August 31, 2015.
- Copyright© 2015 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
Jump to section
- Article
- Abstract
- Control of Angiogenesis by Thalidomide and Its Analogues
- The mTOR Signalling Pathway Might Integrate with Thalidomide Function
- Suppression of Cell Proliferation and Tumour Growth by Thalidomides
- Immunomodulation by Thalidomides
- Clinical Evaluation of Thalidomide and Derivatives
- Concluding Thoughts
- Acknowledgements
- References
- Figures & Data
- Info & Metrics