 Gustavo P. Amarante-Mendes1,2*
Gustavo P. Amarante-Mendes1,2* Sandy Adjemian3,4
Sandy Adjemian3,4 Laura Migliari Branco5,6
Laura Migliari Branco5,6 Larissa C. Zanetti7
Larissa C. Zanetti7 Ricardo Weinlich7*
Ricardo Weinlich7* Karina R. Bortoluci5,6*
Karina R. Bortoluci5,6*- 1Departamento de Imunologia, Instituto de Ciências Biomédicas, Universidade de São Paulo, São Paulo, Brazil
- 2Instituto de Investigação em Imunologia, Instituto Nacional de Ciência e Tecnologia (INCT), São Paulo, Brazil
- 3Molecular Signaling and Cell Death Unit, Inflammation Research Center, VIB, Ghent, Belgium
- 4Department of Biomedical Molecular Biology, Ghent University, Ghent, Belgium
- 5Departamento de Ciências Biológicas, Universidade Federal de São Paulo, Diadema, Brazil
- 6Centro de Terapia Celular e Molecular (CTC-Mol), Universidade Federal de São Paulo, São Paulo, Brazil
- 7Instituto Israelita de Ensino e Pesquisa, Hospital Israelita Albert Einstein, São Paulo, Brazil
Pattern Recognition Receptors (PRRs) are proteins capable of recognizing molecules frequently found in pathogens (the so-called Pathogen-Associated Molecular Patterns—PAMPs), or molecules released by damaged cells (the Damage-Associated Molecular Patterns—DAMPs). They emerged phylogenetically prior to the appearance of the adaptive immunity and, therefore, are considered part of the innate immune system. Signals derived from the engagement of PRRs on the immune cells activate microbicidal and pro-inflammatory responses required to eliminate or, at least, to contain infectious agents. Molecularly controlled forms of cell death are also part of a very ancestral mechanism involved in key aspects of the physiology of multicellular organism, including the elimination of unwanted, damaged or infected cells. Interestingly, each form of cell death has its particular effect on inflammation and on the development of innate and adaptive immune responses. In this review article, we discuss some aspects of the molecular interplay between the cell death machinery and signals initiated by the activation of PRRs by PAMPs and DAMPs.
Introduction
In 1989, Charles Janeway Jr. proposed the existence of a collection of receptors expressed by innate immune cells responsible for detecting conserved products of microbial origin (1). After 25 years of intense research, fierce debates, and a Nobel Prize granted on this subject, it is unquestionable that Janeway's ingenious idea has revolutionized our understanding of the immune system. Indeed, his seminal article is considered as one of the pillars of immunology (2).
The so-called Pattern Recognition Receptors (PRRs) are proteins capable of recognizing molecules frequently associated with pathogens (aka Pathogen-Associated Molecular Patterns—PAMPs). A more comprehensive description of PRRs and their signaling transduction pathways can be found elsewhere (3). Briefly, PRRs can be found associated to subcellular compartments, such as the cellular and endosomal membranes, the cytosol, as well as extracellularly, in secreted forms present in the bloodstream and interstitial fluids (3). There are four major sub-families of PRRs—the Toll-like receptors (TLRs), the nucleotide-binding oligomerization domain (NOD)- Leucin Rich Repeats (LRR)-containing receptors (NLR), the retinoic acid-inducible gene 1 (RIG-1) -like receptors (RLR; aka RIG-1-like helicases—RLH), and the C-type lectin receptors (CLRs) (4). As predicted by Janeway, the engagement of PRRs on the innate immune cells induces co-stimulatory signals for the adaptive immune cells (particularly T lymphocytes) (5). In addition, they activate microbicidal and pro-inflammatory responses required to eliminate (or at least to contain) infectious agents, including the induction of infected cell death (6), as discussed below.
Another ingenious idea came from Polly Matzinger (7), who proposed that the immune system is less concerned with the origin of the antigens (self vs. non-self) than with the context of their encounter with our body (tissue damage vs. tissue homeostasis). In her “Danger Theory,” Matzinger suggested that during tissue stress or damage, endogenous molecules are released or activated and initiate or propagate the inflammatory response, which, among other things, empower antigen-presenting cells to activate the adaptive immune response. Today, these molecules are collectively known as DAMPs (Damage-Associated Molecular Patterns). Importantly, soon enough it became clear that similarly to PAMPs, DAMPs could also engage PRRs.
These two theories together put forward the idea that our body is equipped to distinguish “healthy,” homeostatic tissue turnover or encounters with foreign “friendly” microorganisms, from potential “danger” that may come from pathogens and/or tissue damage.
Cell Death Programs
Molecularly controlled forms of cell death are part of a very ancestral mechanism involved in key aspects of the physiology of multicellular organism, including the elimination of unwanted, damaged or infected cells. Importantly to our discussion, cell death can have a direct or an indirect impact upon the course of infection, as the elimination of infected cells may eradicate or at least restrain the growth of a given pathogen. Moreover, the recognition of dying cells or their by-products modulates both inflammatory and immune responses. In the following sections, we will briefly describe the mechanisms that govern the three major types of molecularly controlled forms of cell death, namely apoptosis, necroptosis and pyroptosis, that participate in host defense through elimination of infected cells, and how they are regulated by signals derived from PRRs. For information regarding other cell death modes please refer to the work published by the Nomenclature on Cell Death Committee 2018 (8)
Apoptosis
Apoptosis was the first type of programmed cell death to be described, initially based on morphological features that distinguished it from necrosis, an uncontrolled, accidental form of cell death observed upon extreme physicochemical insults (9). In this regard, apoptosis is characterized by chromatin condensation, nuclear fragmentation, cell shrinkage with formation of cellular membrane blebs, and, finally, cellular disintegration into fragments known as apoptotic bodies (10). Importantly, during apoptosis, the plasma membrane integrity is preserved, avoiding the release of intracellular contents to the extracellular milieu. This feature contributes to the concept that apoptosis is an (relatively) inflammatory-silent form of cell death. Indeed, recognition and elimination of apoptotic cells during physiological circumstances, such as tissue/organ sculpture during development and tissue homeostasis, occurs without the cardinal signs of inflammation. In addition, it is well established that recognition of apoptotic cells by macrophages, in particular, results in the production of anti-inflammatory molecules, such as TGF-β and PGE2 (11). On the other hand, it is also known that apoptotic cells release a series of so-called “find-me” signals, such as extracellular ATP and lysophosphatidylcholine (LPC), capable of recruiting phagocytes to the site of apoptotic corpses, characterizing, therefore, at least one aspect of an inflammatory reaction (12, 13). Besides, more recently, it was shown that apoptosis initiated via the FAS/CD95 death receptor is associated with the release of chemokines and other immunologically active proteins that coordinates the migration of phagocytes and proper removal of apoptotic cells (14). Taken together, it is reasonable to say that although not completely “silent,” apoptosis is a form of cell death that does not trigger an overt inflammatory response.
From the molecular point of view, much of our knowledge about the regulation of apoptosis came from works with the nematode Caenorhabditis elegans. In a series of elegant studies, Bob Horvitz and colleagues identified four crucial genes (Ced-3, Ced-4, Ced-9, and Egl-1) responsible for the control of developmental cell death in C. elegans (15), which granted him the Nobel Prize in Physiology or Medicine in 2002, together with John Sulston and Sidney Brenner, “for their discoveries concerning genetic regulation of organ development and programmed cell death.” Soon after Horvitz discoveries, it became clear that cell death in C. elegans and apoptosis in mammals shared a very similar, phylogenetically conserved mechanism. Apoptosis is executed by certain members of a family of cysteine aspartate-specific proteases called caspases (16–18). Importantly, not all caspases induces apoptosis. Caspases-1, -4, -5, -11, -12, -13, and-14 are inflammatory caspases not related to the initiation or execution of the apoptotic program. Caspases are produced as an inactive pro-form (zymogen) that can be activated either through proteolytic processing by upstream caspases (in the case of caspases-3, -6, and-7) or via dimerization in the context of multimolecular platforms, such as the apoptosome (caspase-9), the DISC (death-inducing signaling complex) (caspases-8 and-10), the PIDDosome (caspase-2), and the inflammasome (caspase-1 and-11) (16). Executioner or effector caspases, such as caspase-3, -6, and-7 (and CED-3 in C. elegans), are responsible for the induction of the morphological as well as the biochemical features associated with apoptosis, including oligonucleosomal DNA fragmentation and externalization of phosphatidylserine (PS) residues from the inner to the outer leaflet of the plasma membrane (19). Interestingly, in mammals, although the inhibition of effector caspases prevents apoptosis, it does not preclude cell death, which proceeds with different morphological and biochemical characteristics (20). Because of this, it has been proposed that apoptosis in mammals may not be actually a cell death mechanism, but perhaps a termination step of a cell-death program aimed to properly dispose damaged or unwanted cells without initiating inflammatory responses (18).
There are two signaling pathways of apoptosis (Figure 1). The intrinsic pathway deals with signals derived from intracellular stress, such as DNA damage, oxidative stress, dysregulation of Ca2+ homeostasis, interference with the cytoskeleton structure, endoplasmic reticulum stress, etc. Its first layer of regulation comprises the differential expression/activation of BCL-2 family members, responsible for controlling the mitochondria outer membrane permeabilization (MOMP) (21). When the pro-apoptotic stress is too strong for a given cell, MOMP allows the selective release of certain mitochondrial proteins, such as SMAC (second mitochondria-derived activator of caspases)/Diablo (direct IAP binding protein with low pI), HtrA2 (high temperature requirement protein A2)/Omi, and cytochrome c to the cytosol. Cytochrome c associates with APAF-1 (apoptosis-activating factor-1), the mammalian CED-4 homolog, and pro-caspase-9, thereby assembling the apoptosome and enabling caspase-9 to activate the downstream effector caspases. SMAC/Diablo and HtrA2/Omi facilitate apoptosis by preventing the inhibitory action of the inhibitors of apoptosis proteins (IAPs) on the effector caspases. The extrinsic pathway, in comparison, is initiated by the interaction of trimeric, extracellular ligands (TNF-α, CD95L, and TRAIL) to their cognate receptors (TNFR1, CD95 and TRAILRI, or TRAILRII, respectively) present on the plasma membrane (10, 22, 23). The stimulation of these so-called death receptors (DRs) leads to the recruitment of adaptor molecules, such as TRADD (Tumor necrosis factor receptor type 1-associated death domain protein) and/or FADD (Fas-associated protein with death domain), and the pro-caspase-8, giving rise to the conventional DISC. Next, caspase-8 directly activates the effector caspases or amplifies the cell death signal by engaging BID (BH3 interacting-domain death agonist), a pro-apoptotic member of the BCL-2 (B-cell lymphoma 2) family, leading to MOMP, cytochrome c release and assembly of the apoptosome (Figure 1). It is important to mention that the activation of caspase-8 in the context of DISC can be regulated by c-FLIP (cellular FLICE-like inhibitory protein), a catalytically-dead caspase-8 homolog (24).
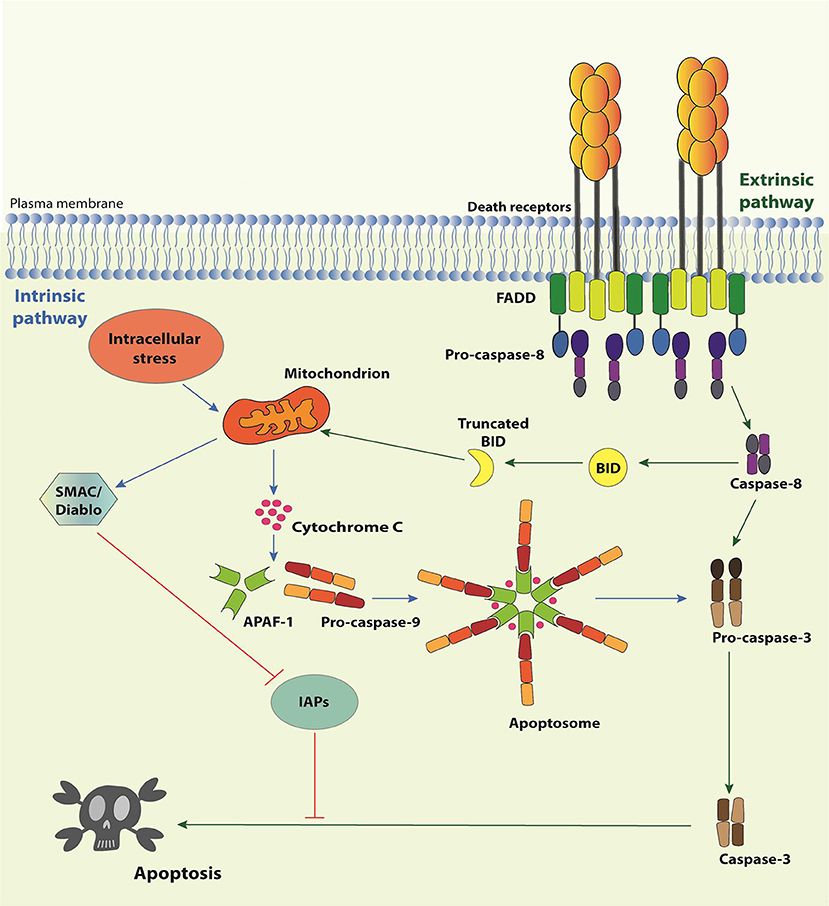
Figure 1. Apoptosis pathways. The Intrinsic Pathway of apoptosis is activated when intracellular “stresses,” such as DNA or cytoskeleton damage or absence of growth/survival factors, are “perceived” by BH3-only members of the Bcl-2 family. These molecules become activated and migrate to the mitochondria where they facilitate or actively induce the release of apoptogenic factors, such as cytochrome c and SMAC/Diablo, to the cytosol. Cytochrome c associates with APAF-1 and pro-caspase-9 to form the apoptosome, resulting in the activation of caspase-9, which activates the effector caspases-3, -6, and -7, responsible for the biochemical and morphological modifications associated to apoptosis. SMAC/Diablo participates by preventing inhibition of caspases by IAPs. The extrinsic pathway of apoptosis initiates by the engagement of Death Receptor by their cognate Death Receptor Ligands causing the formation of the Death-inducing signaling complex (DISC). DISC is formed by the intracellular portion of the Death Receptors, the adaptor proteins TRADD and/or FADD and the pro-caspase-8 (or pro-caspase-10). Activated caspase-8 may directly activate the effector caspases or process the BH3-only protein Bid. Truncated Bid migrates to mitochondria and activates the extrinsic pathway of apoptosis.
In some instances, apoptosis can also be triggered by TLR stimulation, as a defense mechanism against infection. TLR2 was the first PRR to be associated with induction of apoptosis, by virtue of its ability to recruit FADD via MyD88 (Myeloid differentiation primary response 88), and the consequent activation of caspase-8 (25). Likewise, bacterial lipoproteins were reported to trigger apoptosis through this TLR2 pathway (26, 27) and Mycobacterium tuberculosis was also shown to induce TLR-2/caspase-8-dependent apoptosis in macrophages (28). Interestingly, TLR3-induced apoptosis is mediated via TRIF (TIR-domain-containing adapter-inducing interferon-β), which interacts with RIPK1 (Receptor Interacting Serine/Threonine Kinase 1) through its RHIM (RIP homotypic interaction motif) domain (please refer to necroptosis section for further information on these protein-protein interactions). FADD is then recruited, and activates caspase-8 leading to apoptosis (25, 29). In human keratinocytes, poly I:C-induced apoptosis required the stimulation of TLR3 and its adaptor TRIF, thus inducing caspase-8 activation (30); the same molecules were shown to induce apoptosis in human breast cancer cells (31). Not surprisingly, TLR4 can induce apoptosis either via MyD88 or TRIF, and depending on the cell type or conditions engage the extrinsic or intrinsic pathways. For instance, Yersinia was shown to induce TLR4-mediated apoptosis of macrophages through TRIF (32, 33). TRIF-mediated apoptosis seems to be executed through the extrinsic pathway, with no evidence of the involvement of the mitochondrial pathway (34). Interestingly, UV irradiation was shown to induce apoptosis in murine macrophages through TLR4 and MyD88 (35). Despite these observations and a number of other examples that we have not presented here, it is important to emphasize that PRR-induced apoptosis is a relatively minor event compared to all other triggers of apoptosis and that PRR activation leads preferentially to other forms of regulated cell death, as we will discuss below.
Necroptosis
Evidence of a molecularly controlled necrotic cell death was first provided by studies showing that Tumor Necrosis Factor Receptor 1 (TNFR1) and CD95 ligation were capable of inducing necrosis, particularly when caspase activity was inhibited (36, 37). This idea was further supported by a study that demonstrated that the cowpox virus could induce necrosis in porcine kidney cells when it harbored the caspase inhibitor CrmA (cytokine response modifier A) (38). This cell death mode was named “Necroptosis,” as it reflects the existence of a molecular pathway (like apoptosis) but with a necrotic phenotype.
The first molecule to be identified in the necroptotic pathway was RIPK1 as its kinase activity inhibitor, necrostatin-1 (Nec-1), was shown to suppress cell death triggered by caspase inhibition during TNFR1/Fas stimulation (39). RIPK1 has been previously involved in apoptotic and survival pathways, functioning as a scaffold protein to the assembly of the respective signaling platforms (40). Contrastingly, the RIPK1 kinase activity is indispensible for death receptor-triggered necroptosis, as its auto-phosphorylation induces a conformational change that allows RIPK1 to recruit, via their respective RHIM domains, the next member of this pathway, namely RIPK3 (41–43). Once recruited, RIPK3 gets activated by auto-phosphorylation and forms an amyloid-like structure, which promotes the recruitment and activation of Mixed Lineage Kinase Domain-Like (MLKL) (42, 44–47). RIPK3-phosphorylated MLKL oligomerizes and translocates to the plasma membrane, where it interacts with phosphatidylinositides and induces plasma membrane disruption [(48–51); Figure 2]. Distinct effector mechanisms were raised to account for the MLKL-driven permeabilization of the plasma membrane, either directly by pore or cation channel formation, or indirectly, by activation of TRPM or other ion channels (48–52). It is still unclear, however, which of these mechanisms are physiologically relevant. Nonetheless, in all cases, MLKL induces a loss of osmolality control, which causes cell swelling and membrane rupture. Recently, ESCRT-III machinery was suggested to counter these effects by shedding out the MLKL-damaged plasma membrane regions (53).
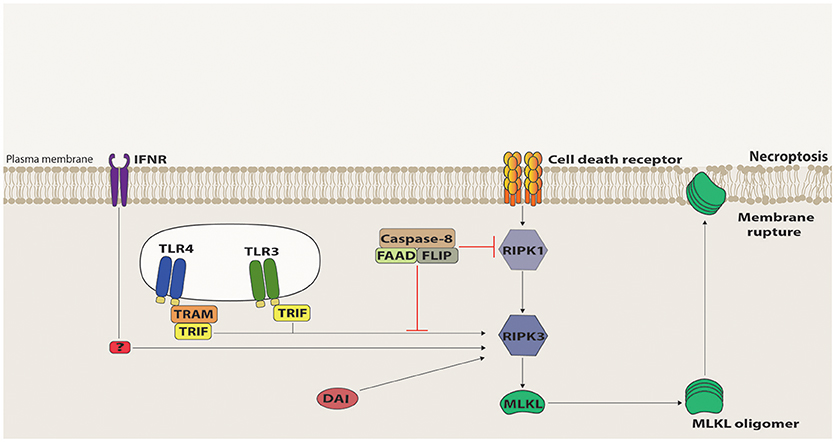
Figure 2. Necroptosis signaling. Death Receptor (DR)-induced necroptosis requires RIPK1 kinase activity to recruit RIPK3 that, in turn, recruits and activates MLKL via phosphorylation of its pseudokinase domain. Once phosphorylated, MLKL oligomerizes and migrates to the plasma membrane, where it interacts with phosphatidylinositol phosphates and induces membrane destabilization and rupture. Necroptosis signaling mediated by TRIF, IFNR, and DAI can directly activate RIPK3 and, in this case, RIPK1 acts as a negative regulator, mostly by recruiting to the signaling platform the suppressive complex containing Caspase-8, FADD and c-FLIP.
Necroptosis can be initiated by a variety of signals. The first to be described and most thoroughly studied was TNFR1 ligation (36). Upon its ligation, TNFR1 typically assembles a multimolecular complex (Complex I) composed by TRADD, RIPK1, TRAF2, TRAF5, cIAP1, cIAP2, and LUBAC (linear ubiquitin chain assembly complex), which is involved in NF-κB activation, pro-inflammatory cytokines synthesis and cell survival (54). Sustained TNFRI ligation leads to CYLD-mediated deubiquitination of this complex, which disassembles, allowing the formation of a secondary complex (Complex II) in the cytosol, constituted by TRADD, FADD, RIPK1, caspase-8, and occasionally c-FLIP (54). As pointed out above, when c-FLIP levels are low, caspase-8 forms active homodimers and triggers downstream events that culminate in apoptosis. However, in the absence of FADD, c-FLIP or a functional caspase-8, TNFRI signaling results in the recruitment of TRADD and RIPK1, forming a platform called complex IIb or necrosome, wherein RIPK3 and MLKL are activated to execute necroptosis (55). Although slightly differing on how RIPK1 is brought to the complex, this molecule has also a central role in Fas and TRAILR-induced necroptosis, as RIPK1 is, in all these cases, mandatory to recruit RIPK3 via their RHIM homotypic domain interactions (54).
Necroptosis can also be triggered by PRRs, such as TLR3 and TLR4, intracellular sensing proteins, such as DAI, RIG-I and MDA-5 as well as interferon signaling [(56–59); Figure 2]. Intriguingly, however, RIPK1 is dispensable for or even inhibitory of the necrosome formation during TLR3-, TLR4-, DAI-, and interferon-mediated necroptosis (57, 60). In these cases, RIPK3 is directly recruited to the signaling platforms, and the presence of RIPK1 slows down or halts the RIPK3-mediated activation of MLKL (60, 61). The ability of RIPK1 to recruit FADD, and consequently, caspase-8 and FLIP accounts, at least in part, for its inhibitory property. Therefore, from the molecular point of view, necroptosis ought to be defined as a RIPK3-dependent form of cell death.
Many other stimuli have been described as capable to induce necroptosis, ranging from UV irradiation, chemotherapeutic drugs (such as cisplatin, etoposide, and staurosporine), natural compounds (such as shikonin and its analogs), to DNA damage, hypoxia, ischemia/reperfusion and oxidative stress (62). The signaling pathways that lead to necroptosis in each of these cases are still to be fully elucidated. Further studies are required to evaluate whether they are dependent on RIPK1 and also whether they directly signal to a RIPK3-activating platform or indirectly, via up regulation of a classic necroptotic inducer, such as TNF or FasL. For example, UV irradiation was reported to induce necroptosis via TNF upregulation but also via spontaneous aggregation of RIPK1 and RIPK3, independently of any death receptor ligation (29, 63). Particularly puzzling is the fact that shikonin, a naphthoquinone compound obtained from a plant extract, can induce necroptosis even in the absence of FADD/caspase-8/FLIP inhibition, which is thought to be mandatory for this type of cell death (64). Thus, either this compound can itself somehow block their activity, or it shall be instrumental to decipher alternative ways in which MLKL is activated and necroptosis is executed.
Nonetheless, despite the different mechanisms that initiate necroptosis, in all cases cells undergo rapid MLKL-mediated plasma membrane permeabilization with consequent release of intracellular contents, including many DAMPs, such as lysosomal proteases, DNA, mtDNA, ATP, and HMGB1 [(55); Table 1). Therefore, similarly to pyroptosis (see below), necroptosis is considered a pro-inflammatory form of cell death. Even so, it is still to be determined whether the pro-inflammatory properties of necroptotic cells are the result of the intracellular content leakage or, rather, they can actively produce and/or modify specific DAMPs. Evidence for the latter comes from ESCRT-III-deficient cells that undergo necroptosis much faster, which limits the amount of inflammatory cytokines and chemokines produced and hinders antigen cross-presentation (53). Moreover, both RIPK3 and MLKL have been associated with inflammasome and NF-κB activation, supporting the notion that the pro-inflammatory potential of necroptotic cells goes beyond the passive release of their intracellular content (110–112).
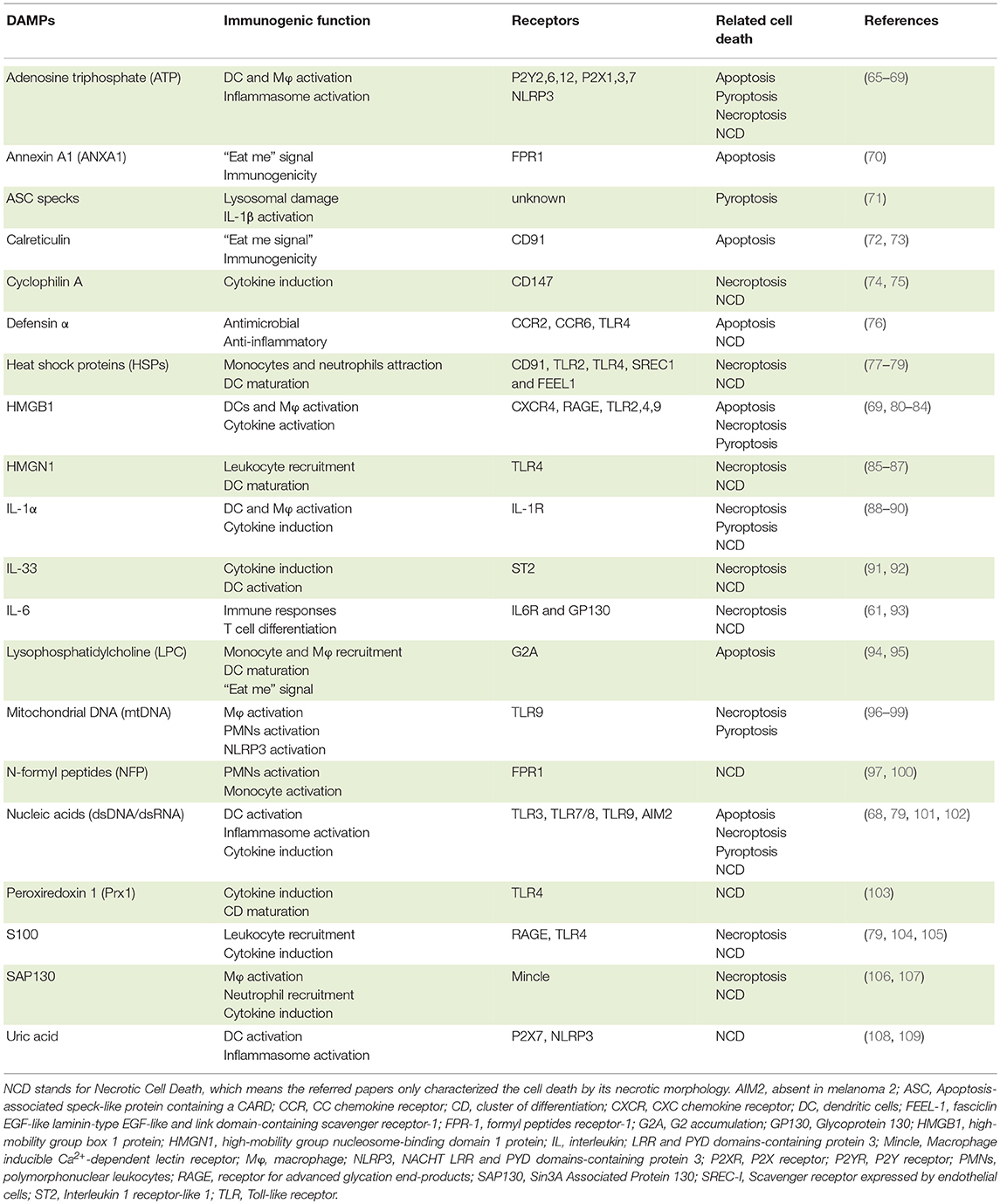
Table 1. DAMPs released by cell death and its role in the immune system.
Necroptotic cells not only induce a potent inflammatory response but they are also highly immunogenic, which may be instrumental against infection and during anti-tumoral responses. For example, mice injected with necroptotic cells present a higher CD8+ T cell cross-priming and increased tumor immunity when compared with animals injected with apoptotic cells (113, 114). Likewise, RIPK3 deficiency in mice inhibits immune cell infiltration and attenuates organ injury during sepsis (115). Therefore, given that necroptosis is highly immunogenic, disruption in the necroptotic pathway would be expected in some pathophysiological conditions. Indeed, it was reported that most of the in vitro transformed cells as well as human tumor samples have low or no expression of RIPK3 (116), and a cohort of chronic lymphocytic leukemia patients present down regulation of CYLD (117). Furthermore, patients with lower expression of RIPK3 or MLKL have worse prognosis for breast cancer or ovarian cancer, respectively (116, 118), suggesting that resistance to necroptosis is positively selected during tumor growth and/or development. This may be associated with an increased ability to evade immune attack, either by prolonging the lifespan of the transformed cells, by decreasing the availability of DAMPs, or by avoiding the activation of antigen-presenting cells during the immune responses. Therefore, induction of necroptosis in tumors may change its immunogenicity and promote a better immune response against it. This is particularly exciting, as we are currently witnessing novel and promising approaches in tumor treatment that are based on stimulation of the immune system. On the other hand, it is possible that the inflammation generated by necroptosis may promote tumor development by stimulating angiogenesis and metastasis (119). Therefore, thorough investigation of the benefits and pitfalls of inducing inflammatory cell death for each cancer type will be required in order to determine whether inducing necroptosis is indeed a good option in the specific cancer treatment.
Besides its impact on tumorigenesis and tumor progression, deficient necroptotic signaling can be detrimental during viral infection. Mice lacking RIPK3 are highly sensitive to vaccinia virus due to widespread infection (120). Likewise, RIPK3-deficient mice are more susceptible to Influenza A virus (IAV) than the wild-type animals (121). Remarkably, seasonal IAV, but not the 1918 and 2009 pandemic IAV strains, induces RIPK3-mediated immunogenic death of dendritic cells (122). The pandemic strains' ability to suppress necroptosis was mapped to the hemagglutinin (HA) genomic segment (122), indicating that either the pandemic strains' HA do not induce necroptosis or it may directly interfere with the necroptotic signaling pathway.
Keeping with the notion that suppressing necroptosis is advantageous to the infectious agent, there is accumulating evidence that viruses can encode molecules that are able to directly interfere with the necroptotic signaling. vIRA, a molecule expressed by MCMV that contains a RHIM-like domain blocks RIPK3 recruitment to RIPK1 and to DAI (57). MCMV expressing vIRA mutated in its RHIM domain produces an attenuated viremia in wild-type mice, which is reverted in RIPK3-deficient animals (57). Likewise, HSV-1 and HSV-2 express ICP-6 and ICP-10, respectively, which are able to suppress necroptosis in human cells through a similar RHIM-dependent mechanism (123, 124). Curiously, in mice, ICP-6 was shown to promote necroptosis through direct aggregation with RIPK3, restricting virus propagation (124, 125). A different mode of action was reported for the IE1-regulated gene product expressed by HCMV, which suppresses necroptosis downstream of RIPK3 activation and MLKL recruitment (126).
Bacteria can also induce necroptosis, at least in vitro. It is less clear, though, whether necroptosis plays a central role in bacterial infections in vivo. Loss of RIPK3 in combination with deletion or inhibition of caspase-8 or FADD renders mice susceptible to a number of pathogens, including Yersinia and Citrobacter (127, 128). However, the relative contribution of necroptosis and caspase-8-mediated apoptosis in these models were not yet tested, as caspase-8- or FADD–deficient animals are not viable (129–131).
Necroptosis, though, may not always be protective against infection. Macrophage death by necroptosis correlates with increased susceptibility to Salmonella infection (132). Also, HIV-specific CD8+ T cell response, which is a key indicator of infection control, is impaired due to increased necroptosis levels in this cell population (133). Taken together, necroptosis seems to be detrimental when it eliminates the population that is central for the immune control of the infection. In the other cases, necroptosis limits infection, mostly likely by destroying the pathogen's replicative niche through a cell death mode that generates a pro-inflammatory and immunogenic environment. However, it is important to note that, as mentioned above, RIPK3 and MLKL were shown to participate of additional signaling platforms, including inflammasome activation, NF-κB signaling and even apoptosis induction (134). Therefore, in the light of these novel RIPK3 and MLKL roles, it is essential to reevaluate the relative contribution of necroptosis to the phenotypes observed. A good illustration comes from the fact that while RIPK3-deficient mice are more susceptible to IAV, MLKL-deficient animals are not, indicating that necroptosis is not the sole RIPK3-mediated mechanism important in IAV control (121). In fact, it was shown that IAV also triggers RIPK3-mediated apoptosis, via recruitment of RIPK1, FADD and caspase-8. This was further supported by the fact that MLKL-caspase-8 double deficient mice present similar levels of susceptibility to IAV infection observed with the RIPK3-deficient animals (121). Another example is that RIPK3-deficient mice are less susceptible to Staphylococcus aureus lung damage and present reduced bacterial loads and inflammation, while MLKL-deficient animals present an opposite outcome, suggesting that these molecules have independent, non-necroptotic roles (135).
Pyroptosis
Pyroptosis is a necrotic form of regulated cell death distinct from necroptosis, mainly due to the requirement of inflammatory caspase-1 and/or caspase-11 (murine caspase-11 corresponds to caspases-4 and -5 in humans) [(136); Figure 3). It is the result of pore formation in the plasma membrane that increases osmotic pressure ensuing in osmotic lysis and, consequently, the release of the intracellular content, including pro-inflammatory cytokines and DAMPs (137). Although distinct from the typical oligonucleosomal fragmentation observed during apoptosis, DNA fragmentation is also a hallmark of pyroptosis, which seems to occur independently of the caspase-activated DNase (CAD) (138).
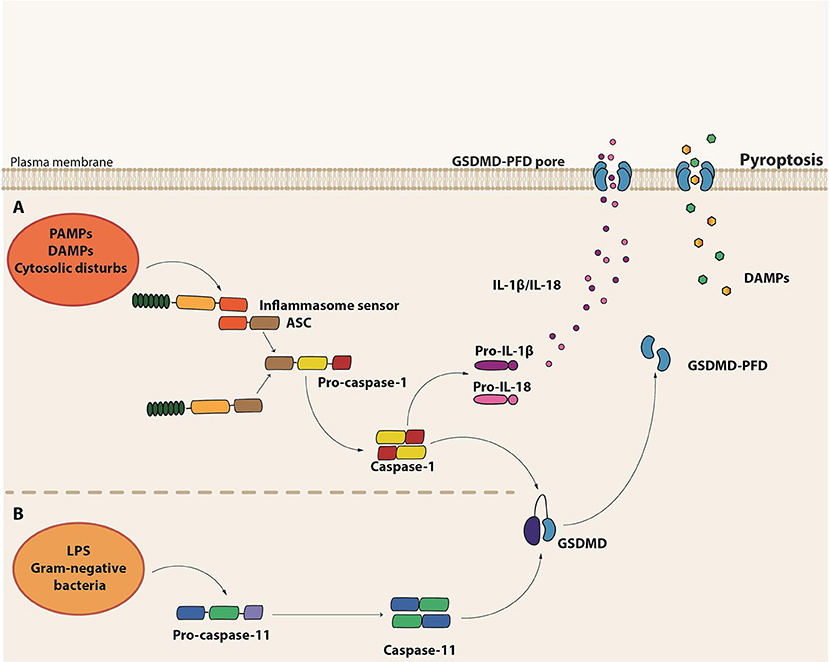
Figure 3. Molecular basis of pyroptosis. (A) Canonical inflammasome assembly upon sensing of PAMPs, DAMPs or other cytosolic disturbs leads to the recruitment and activation of caspase-1 directly or via the recruitment of the adaptor protein ASC. Caspase-1 induces the maturation of pro-IL-1β and pro-IL-18 into their active forms as well as cleavage of Gasdermin D (GSDMD). The GSDMD pore form domain (PFD) interacts with the plasma membrane to form the GSDMD pore, leading to the release of the intracellular content, including IL-1β and IL-18. (B) Non-canonical inflammasome activation is initiated by the detection of cytosolic LPS from gram-negative bacteria by the pro-caspase-11 itself. Activated caspase-11 (caspase-4 or caspase-5 in humans), in turn, induces GSDMD cleavage and consequent pyroptosis.
Pyroptosis is a form of cell death initiated in response to the engagement of certain members of the PRRs, which are capable of assembling complex structures called inflammasomes. These platforms are composed by a sensor protein, either from the NLR or the pyrin and HIN domain-containing protein (PYHIN) families of cytosolic PRRs, in addition to the adaptor molecule apoptosis-associated speck-like protein containing a caspase activating and recruitment domain (ASC) and pro-caspase-1. There are five major types of the so-called canonical inflammasomes—NLR pyrin domain-containing 3 (NLRP3), NLRP1, neuronal apoptosis inhibitory protein (NAIP)/NLR CARD-containing 4 (NLRC4), absent in melanoma 2 (AIM2), and PYRIN inflammasomes (139). Activation of one of these cytosolic sensors in response to PAMPs, DAMPs or cytosolic disturbances such as ionic imbalance leads to the recruitment and activation of caspase-1 either directly or through the ASC adaptor molecule [(140); Figure 3]. Besides the induction of pyroptosis, caspase-1 also leads to the processing and release of the pro-inflammatory cytokines IL-1β and IL-18 (139).
In contrast to the canonical inflammasomes, which require multicomplex structures, the non-canonical inflammasome seems to be composed solely by pro-caspase-11, which plays the role of the sensor as well as the executor (141, 142). During intracellular gram-negative bacteria infections, Lipid A, a component of LPS, can directly bind to the CARD domain of pro-caspase-11 (143), which gets activated and induces pyroptosis. Interestingly, the non-canonical caspase-11 inflammasome acts independently of LPS recognition by TLR4 and does not directly induce IL1-β and IL-18 maturation [(141, 142); Figure 3]. In monocytes, however, non-canonical inflammasome stimulation may result in minor production of IL-1β and IL-18 through the bystander induction of NLRP3 activation (144). Interestingly, LPS-induced lethal shock is driven by the activation of the non-canonical inflammasome. Since IL-1β and IL-18 release are not a major outcome of caspase-11 activation, the exacerbated inflammatory response observed in sepsis seems to be mainly driven by pyroptosis, probably due to the efflux of DAMPs, such as High Mobility Group Box 1 (HMGB1) and IL-1α [Table 1; (145)].
Such non-canonical inflammasome-mediated responses have drawn the attention of different research groups that became interested in unraveling the relevant as well as the pathogenic caspase-11 downstream targets. In 2015, two concurrent studies reported that Gasdermin D (GSDMD), a member of the GSDM family, was the effector component of the non-canonical inflammasome pathway (146, 147), which was later confirmed by a third study (148). Kayagaki and colleagues performed N-ethyl-N-nitrosourea mutagenesis screening for mutations that compromised LPS-induced IL-1β release and pyroptosis (146) while Shi and colleagues employed the clustered regularly interspaced palindromic repeat (CRISPR)/Cas9 genome-editing screens in TLR4 deficient mouse bone marrow-derived macrophages for guide RNAs that protected from LPS-induced cell death (147). Both studies hit GSDMD as a substrate for caspase-11 and the effector of pyroptosis. GSDMD is composed by a C-terminal and a N-terminal domain linked by a long loop. Caspase-11 cleaves an aspartate residue within the linking loop, releasing the N-terminal fragment from the inhibitory C-terminus (146, 147). The N-terminal domain, also called Pore-Forming Domain (PFD) (149) oligomerizes and associates with lipids in the inner plasma membrane to form 10–33 nm pores leading to cell swelling and eventually to cell lysis (150–153). Importantly, it was also demonstrated that caspase-1 cleaves GSDMD at the same site as caspase-11, establishing that GSDMD is also required for the canonical inflammasome-driven pyroptosis (147).
Until the discovery of GSDMD as the pyroptosis executioner, the physiological function of GSDM proteins was largely unknown. However, recent studies described that the PFD is highly conserved among several members the GSMD family. Indeed, expression of PFD from GSDMA, GSDMA3, GSDMB, GSDMC, GSDME, or GSDMA3 in HEK293 was able to induce pore formation and a cell death phenotype similar to pyroptosis (147, 151). Moreover, GSDMA3 cleavage by caspase-3 in HEK293 and macrophages results in a secondary necrotic cell death after apoptosis (154). This necrotic cell death might contribute to hearing loss in GSDMA3 spontaneous mutations that are associated with deafness (155). Thus, given the cytotoxic activity of different GSDM PFD, some authors have proposed a redefinition of pyroptosis as a GSDM-mediated cell death (146). However, it is controversial how other GSDM members are activated and whether these proteins participate in cell death pathways. Also, GSDMD seems not to be required for pyroptosis during prolonged inflammasome activation in response to the classical agonists, ATP, and flagellin (146). Moreover, in the absence of caspase-1 protease activity, caspase-8 accounts for GSDMD-independent cell death in response to inflammasome agonists (156–158). Since some of these processes share features of pyroptosis, it is hard to define pyroptosis solely as being a process of cell death regulated by inflammatory caspases or mediated by GSDM proteins, since we can find exceptions to the rules that govern both concepts.
From the biological point-of-view, cell death by pyroptosis results in a fast removal of infected cell leading to the elimination of the replication niche. Conversely to the previous idea of liberation of bacteria to the extracellular milieu by pyroptotic cells (159), the current knowledge predicts that, instead, the damaged bacteria remain trapped within the pyroptotic corpses. This structure is called pore-induced trap (PIT) and it prevents bacterial dissemination (160, 161). Despite that PIT does not directly kill intracellular bacteria, pyroptosis renders them more susceptible to H202, to the antimicrobial peptide polymyxin B and to the antibiotic ciprofloxacin (157). As a consequence, the recovered bacteria from PIT are less capable to infect neighbor cells.
The inflammatory milieu created by the release of the intracellular content from pyroptotic cells recruits circulating phagocytes to the infectious site. Subsequently, neutrophils efferocyte the PIT and kill the pathogen by a mechanism dependent on reactive oxygen species (ROS) (161). Extracellular bacteria can also be controlled by the action of antimicrobial peptides (160, 161) and potentially by the GSDMD N-terminal domain released during cell lysis due to its affinity to cardiolipin and phosphatidylserine expressed in some bacterial cell membranes, such as Escherichia coli and Listeria monocytogenes (152, 162). Interestingly, canonical and non-canonical inflammasomes are required for intestinal epithelial cells (IECs) responses to infections (163, 164). The activation of NLRC4 inflammasomes in IECs results in a lytic cell death prior to a non-conventional process of cell expulsion that contributes to control bacterial replication. Although caspase-1 and Gasdermin-D were required for IEC pyroptosis, both molecules were dispensable for cell expulsion, demonstrating that coordinated inflammasome responses in IECs are important to prevent bacterial translocation to deeper tissues (163, 164).
Interestingly, neutrophils seems to be more resistant to pyroptosis than macrophages in response to Salmonella and are able to maintain a sustained IL-1β production and secretion, which could be important to control the infection (165). However, Kambara et al. (166) recently described that a specific neutrophil elastase (ELANE) is able to cleave GSDMD independently of caspases activity, promoting a lytic cell death in these cells. Interestingly, these authors demonstrated that GSDMD-dependent neutrophil death impairs the control of extracellular bacteria E. coli, thus suggesting that GSDMD could exert an anti-inflammatory role depending on the infection context.
In addition to its role in the elimination of replicative niche, the pyroptosis machinery is involved in IL-1β and IL-18 release. As these cytokines lack the signal peptide, their release is considered to occur by non-conventional pathways (167). Among the different pathways that have been proposed to explain their secretion, mechanisms involving cell death are particularly subject to intense debate in the literature. Growing evidences suggest that IL-1β can be released by viable monocytes (168), dendritic cells (DCs) (169), and macrophages (170). GSDMD pore is large enough to allow IL-1β release concomitant with the influx of cationic ions (148). Notably, in viable cells, GSDMD seems to be required for IL-1β translocation to the extracellular space in response to stimuli that hyperactivate phagocytes, such as oxidized phospholipids (oxPAPC) in DCs or LPS in human monocytes (170–172). Nonetheless, it is difficult to establish whether the cells were actually viable, since cell death can precede cell lysis, thus suggesting that pyroptosis and cell lysis can be uncoupled events (173). Moreover, the assessment of cell death by the detection of lactate dehydrogenase release (LDH), used in several studies as the only viability assay, might be insufficient to discriminate viable cells from dying cells since both viable and unviable cells can release LDH to the cell culture (170, 173, 174).
Although many studies have demonstrated the requirement of canonical and non-canonical inflammasomes to host defense against pathogens, the precise contribution of pyroptosis and other inflammasome-related mechanisms are poorly understood and arose mainly from in vitro assays or bacterial infection models in mice deficient for molecules that compose these platforms (159, 175). In L. monocytogenes, S. typhimurium, and B. thailandensis in vivo infections, the lack of caspase-1/11 was more deleterious to the host than IL-1β/IL-18 deficiency (176–179), while in mice infected with F. novicida, the treatment with recombinant IL-1β/IL-18 only partially recovered the resistance to infection, suggesting that cytokine secretion was not sufficient to protect the host (180). The susceptibility of GSDMD-deficient mice seems to correlate with that demonstrated by caspase-1/11-deficient mice, although the deletion of GSDMD also culminated in reduced IL-1β/IL-18 release (146–148). Even though (a) IL-1β/IL-18 secretion might occur independently of pyroptosis and (b) caspase-1 or caspase-11 deletion is more severe than IL-1β/IL-18 ablation in some bacterial infections, usually cytokine release and cell death are overlapping events necessary for optimal host defense (159). In any case, the susceptibility of GSDMD and other GSDM deficient mouse strains to infectious agents and its comparison to mice lacking caspase-1, caspase-11, IL-1β, and/or IL-18 remains to be established, especially in non-bacterial infection contexts. For example, despite clear evidences of the involvement of inflammatory caspases in the host control of some fungal infections such as Candida albicans, Aspergillus fumigatus, and Paracoccidioides brasiliensis (172), the requirement of GSDMD to cell death and the consequences to the host resistance against these infections is still to be elucidated.
Notwithstanding, the highly pro inflammatory outcome of pyroptosis as well as the cell loss can be prejudicial to the host during the response to pathogens. In HIV patients, the quiescent CD4 T cells depletion seems to be mainly mediated by pyroptosis (181, 182). During HIV abortive infection, the engagement of the interferon-gamma-inducible-protein 16 (IFI16) in response to cytosolic viral DNA leads to inflammasome assembly and caspase-1 mediated CD4 T cells pyroptosis in lymphoid tissues (181, 182). Interestingly, co-cultivation of lymphoid-derived cells sensitizes blood-derived CD4 T cells to HIV-induced pyroptosis (183). Moreover, pyroptotic peripheral blood CD14+CD16− monocytes from HIV-infected patients release ASC specks, a hallmark of inflammasome activation. Therefore, besides the depletion of CD4 T cells, pyroptosis of CD4 T cells and monocytes contributes to the chronic inflammation that characterizes the disease (184).
The identification of the non-canonical inflammasome and the discovery of GSDMD as the executioner of pyroptosis have expanded our understanding of the mechanisms driving this type of cell death. However, further studies are necessary to elucidate the precise role of inflammatory and non-inflammatory caspases and the participation of members from GSDM family and/or other effector proteases in the molecular regulation of pyroptosis. In addition, the understanding of its role during infection or inflammatory processes in vivo will contribute to better understand the biological relevance of this regulated cell death induced in response to the PRRs activation.
PRR Sensing of Cell Death and Cell Death Products
The notion that cells undergoing cell death release or expose several intracellular molecules regardless of the accidental nature or the different regulated death programs (apoptosis, necrosis or pyroptosis) is widely recognized. Although mainly non-inflammatory in the intracellular space, molecules released/exposed from damaged cells can participate in the activation of inflammation and immune responses. Indeed, a broad range of receptors, including PRR, sense these DAMPs and alert the immune system by inducing immune cell migration, increasing phagocytosis by macrophages and DCs, stimulating the production of pro-inflammatory cytokines or even contributing to the maturation of DCs, among other key functions (72, 80, 185). A number of studies have been dedicated to the characterization of putative DAMPs, and it became apparent that the type of cell death, as well as the nature of cell death stimuli, influence the quality and quantity of DAMPs release (Table 1).
Importantly, the stress or damage before the cellular demise itself is determinant to set in motion a sequence of events leading to an immunogenic cell death (ICD). The sensing of this stress regulates the cell death process thus initiating signaling pathways that will actively—or not—generate danger signals (186). Other DAMPs will be passively released as a result of membrane rupture during necroptosis or pyroptosis. These DAMPs define in part the immunogenicity of cell death, but are not sufficient to elicit a specific anti-tumor immune response, for instance. Indeed, they are released or exposed by the dying cells and act as adjuvant providing that antigens are exhibited conjointly (187). In contrast, a non-immunogenic cell death does not provide the required levels of DAMPs and antigens to evoke an adaptive immune response (187).
Together, these concepts redefined the widely accepted paradigm stating that apoptosis is always a silent cell death modality as opposed to necrosis, which is inflammatory and immunogenic. Therefore, a non-immunogenic apoptosis is characterized by the absence of plasma membrane leakage and the rapid phagocytosis of apoptotic bodies prevents the release of DAMPs and the consequent inflammatory reaction. Indeed the apoptotic process reduce cell immunogenicity by diverse ways including, (1) preservation of intracellular structures and plasma membrane, thereby blocking the release of DAMPS; (2) reduction of cellular volume, by condensation of the nucleus and shedding of small vesicles, which favors its rapid elimination by the surrounding tissue; (3) expression of “find-me” and “eat-me” signals, which increases the speed of cell clearance (12, 188); (4) inhibition of the production of interferons and pro-inflammatory cytokines, as the DNA is being chopped and condensed (189); (5) induction of an antigenic tolerance in the engulfing APCs (190); and (6) post-translational modifications on DAMPS and alarmins that decrease their pro-inflammatory potential (191, 192).
Interestingly, depending on the trigger, apoptosis can be immunogenic. Indeed, some chemotherapeutic agents, such as anthracyclines, as well as radiation and hypericin-based photodynamic therapy, were found to strongly prime immune responses through the induction of ICD (65, 185). Among these, immunogenic chemotherapies are well characterized and involve the emission of a number of danger signals. The pre-apoptotic release or exposure on the plasma membrane of ER-chaperones, such as calreticulin and Heat Shock Proteins (HSPs), constitutes an early event of ICD, which relies on the induction of an ER-stress. Calreticulin promotes the uptake of dying cells by DCs (72) and the inhibition of its exposure during anthracycline-induced apoptosis of murine tumor cell lines abolished their immunogenic potential (72). The early apoptotic secretion of ATP, which binds to P2X7 or P2Y2 purinergic receptor on DCs, stimulates the formation of the NLRP3 inflammasome, thereby inducing the release of IL-1β (66). Moreover, ATP released by dying cells undergoing ICD is responsible for the recruitment and differentiation of myeloid precursors into inflammatory DCs, mediating a specific antitumor immune response (193). Passive release of the nuclear protein HMGB1 occurs during secondary necrosis (i.e., late-stage apoptotic cells), which interacts with TLR4 on DCs, and through Myd88 signaling, enables efficient tumor antigen processing and cross-presentation (80). Additionally, anthracyclines have been shown to induce the release of RNA, thereby stimulating TLR3 as a mimic of viral infection. Activation of TLR3 is then responsible for type I IFNs production that acts in an autocrine and paracrine manner to promote the secretion of CXCL10 (194). Release of Annexin A1 has also been described after anthracyclines treatment, stimulating the Formyl Peptide Receptor 1 (FPR1), thus directing the final trajectory of DCs to dying tumor cells (195).
Besides chemotherapeutic agents, bacterial and viral infection can also trigger an immunogenic apoptosis. In this case, PAMPs, such as LPS or double-stranded RNA, expressed by the pathogen can stimulate TLR signaling and induce an immune response. Indeed, phagocytosis of infected apoptotic cells could trigger TLR activation and lead to IL-6 and TGF-β production that induce the development of infection-specific as well as self-reactive TH17 cells, linking infection to autoimmunity (196, 197). Finally, defects in mechanisms of apoptotic cell clearance are linked to autoimmunity disorders, including lupus and rheumatoid arthritis, likely due to the increased risk of loss of cell integrity with the consequent release of DAMPs and increased availability of circulating self-antigens (198, 199).
Accidental or programmed forms of necrosis are responsible for the release of an usually larger panel of DAMPs compared to apoptotic cells, mainly due to plasma membrane permeabilization. Also, RIPK1-mediated activation of the NF-κB pathway, through upregulation of pro-inflammatory cytokines and increased antigen presentation, was reported to play a role in the immunogenicity of necroptotic cell death mode (186). Recently, it was show that necroptosis is accompanied by the release of the classical and potent DAMPs—HSPs, ATP, and HMGB1 (200, 201). While HSPs were shown to stimulate TLR2 and 4, inducing DCs maturation, HMGB1 was reported to interact with TLR3, 4 and 9, as well as RAGE, to activate DCs and macrophages (202–204). Necrotic cells can potentially release intact mitochondria, a major source of ATP, which may activate the NLRP3 inflammasome resulting in IL-1β secretion and neutrophil recruitment (67, 96, 205). Moreover, mitochondrial DAMPs, such as formyl peptides and mitochondrial DNA, can potentially act on FPR1 and TLR9, respectively, inducing neutrophils recruitment and degranulation (97, 115). Additionally, Mincle, the C-type lectin receptor 4E was reported to interact with the necrotic DAMP SAP130 (spliceosome-associated protein 130), normally involved in spliceosomes assembly. The stimulation of this PRR was also reported to induce recruitment of neutrophils (106). Uric acid has been described as a product of accidental necrosis (108). Once released, this DAMP can precipitate and form monosodium urate (MSU) crystals able to induce the expression of costimulatory molecules by DCs, as well as activating the NLRP3 inflammasome to trigger the production of IL-1β and IL-18 (206, 207). RNA and double-stranded DNA (dsDNA) are also released during necrotic cell death, and while RNA stimulates TLR3, dsDNA stimulates TLR9 and two members of the RLR family, RIG-I and MDA5, responsible for the release of IFN-β and CXCL10 through IRF3 and NF-κb pathways. The AIM2 inflammasome can sense dsDNA released by necrotic cells, thereby inducing IL-1β secretion (204). Finally, it is important to mention that some proteins considered DAMPs also stimulate receptors that are not PRRs. For instance, IL-1α acts on IL-1R1 to trigger an inflammatory response mediated by the Myd88 pathway (208) and full-length IL-33 is another DAMP released during necrosis, with immunological property due to the absence of caspase processing (191).
Conclusions
The protective response of our body against pathogens and tumor cells depends on proper activation of both innate and adaptive immunity. Particularly, macrophages and DCs reside on the center of these two arms of immunity. They are powerful antigen-presenting cells that may elicit effector T cell responses (protection) or induce T cells to become regulatory (tolerance), depending on their activation status. They express PRRs, which are very ancient proteins that help us identify and react to pathogens and danger signals. Upon engagement, through the interaction with conserved molecular patterns frequently associated with pathogens (PAMPs), PRRs trigger a series of biochemical signaling cascades that activates pro-inflammatory programs on DCs that enable the differentiation of antigen-specific T cells into protective effector TH1, TH2, and TH17 cells. PRR engagement also triggers programs of cell death, particularly necroptosis and pyroptosis, the necrotic forms of cell death associated with a pro-inflammatory outcome (Table 2). These forms of cell death release larger amounts of DAMPs, which in turn, stimulate surrounding cells via PRRs, thus constituting a positive feedback loop capable of amplifying host defense mechanisms (Figure 4). Apoptosis, on the other hand, is a cell death program mostly related to non-inflammatory outcomes and likely to take major role in the maintenance of homeostasis by “silently” eliminating unwanted or damaged cells. However, apoptosis may also participate in elimination of infectious agents or tumor cells. Therefore, recognition of pathogen- and damage/danger-associated molecules by the same set of immune receptors (PRRs) is a powerful strategy that bridges intrinsic cell death programs and complex immune cell interactions to preserve homeostasis and at the same time protects the organism against infection and cellular transformation (Figure 4).
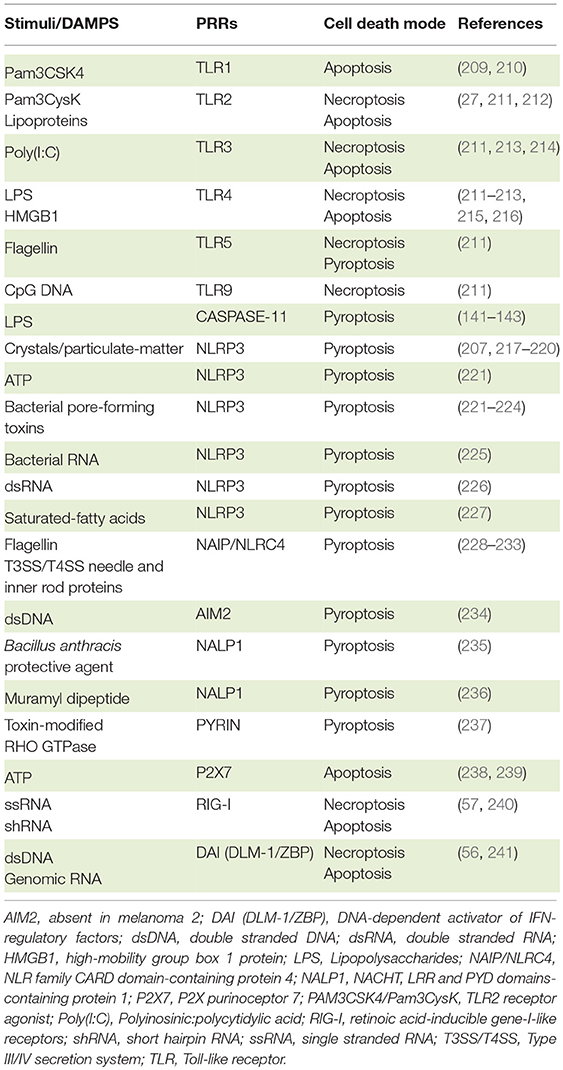
Table 2. PRR agonists and consequent cell death program.
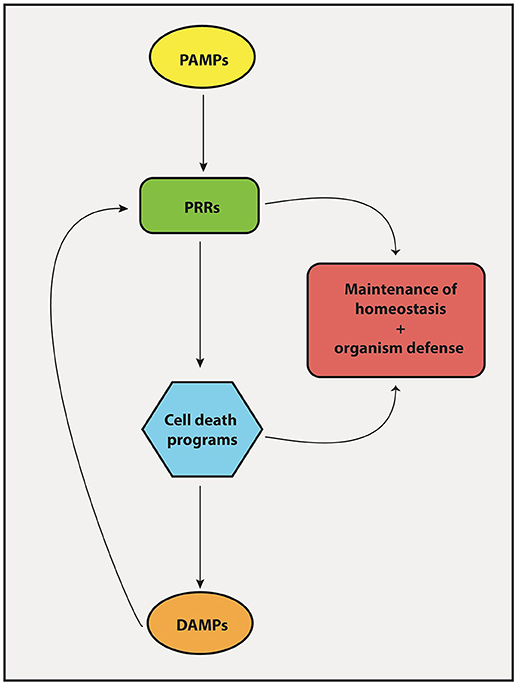
Figure 4. Interplay between PRRs and cell death mechanisms. The engagement of PRRs in response to PAMPs induces the activation of different cell death machineries in order to promote tissue homeostasis and host-defense against pathogens. Importantly, cell death products known collectively as DAMPs forms a feedback loop that stimulate PRRs to induce inflammatory/immune responses.
Author Contributions
LM designed the figures. LZ and SA prepared Table. GA-M, RW, and KB designed the review organization. All authors contributed to the writing, reviewed, and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer FD and handling Editor declared their shared affiliation at the time of review.
Acknowledgments
LM and LZ are recipients of Ph.D. fellowships from FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo, Brazil). SA is recipient of a postdoctoral fellowship from FWO (Fonds voor Wetenschappelijk Onderzoek—Vlaanderen, Belgium). GA-M, KB, and RW are supported by grants from FAPESP and from the Brazilian Research Council (CNPq).
References
1. Janeway CA Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. (1989) 54(Pt 1):1–13. doi: 10.1101/SQB.1989.054.01.003
2. Janeway CA Jr. Pillars article: approaching the asymptote? Evolution and revolution in immunology. Cold spring harb symp quant biol. 1989. 54: 1–13. J Immunol. (2013) 191:4475–4487.
3. Medzhitov R, Janeway CA Jr. Innate immunity: impact on the adaptive immune response. Curr Opin Immunol. (1997) 9:4–9.
4. Walsh D, McCarthy J, O'Driscoll C, Melgar S. Pattern recognition receptors–molecular orchestrators of inflammation in inflammatory bowel disease. Cytokine Growth Factor Rev. (2013) 24:91–104. doi: 10.1016/j.cytogfr.2012.09.003
5. Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature (1997) 388:394–7. doi: 10.1038/41131
6. Bortoluci KR, Medzhitov R. Control of infection by pyroptosis and autophagy: role of TLR and NLR. Cell Mol Life Sci. (2010) 67:1643–51. doi: 10.1007/s00018-010-0335-5
7. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
8. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
9. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer (1972) 26:239–57. doi: 10.1038/bjc.1972.33
10. Amarante-Mendes GP, Green DR. The regulation of apoptotic cell death. Braz J Med Biol Res. (1999) 32:1053–61.
11. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. (1998) 101:890–8. doi: 10.1172/JCI1112
12. Medina CB, Ravichandran KS. Do not let death do us part: ‘find-me' signals in communication between dying cells and the phagocytes. Cell Death Differ. (2016) 23:979–89. doi: 10.1038/cdd.2016.13
13. Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol. (2013) 5:a008748. doi: 10.1101/cshperspect.a008748
14. Cullen SP, Henry CM, Kearney CJ, Logue SE, Feoktistova M, Tynan GA, et al. Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol Cell (2013) 49:1034–48. doi: 10.1016/j.molcel.2013.01.025
15. Metzstein MM, Stanfield GM, Horvitz HR. Genetics of programmed cell death in C. elegans: past, present and future. Trends Genet. (1998) 14:410–6. doi: 10.1016/S0168-9525(98)01573-X
16. Pop C, Salvesen GS. Human caspases: activation, specificity, and regulation. J Biol Chem. (2009) 284:21777–81. doi: 10.1074/jbc.R800084200
17. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. (2013) 5:a008656. doi: 10.1101/cshperspect.a008656
18. Pereira WO, Amarante-Mendes GP. Apoptosis: a programme of cell death or cell disposal? Scand J Immunol. (2011) 73:401–7. doi: 10.1111/j.1365-3083.2011.02513.x
19. Martin SJ, Finucane DM, Amarante-Mendes GP, O'Brien GA, Green DR. Phosphatidylserine externalization during CD95-induced apoptosis of cells and cytoplasts requires ICE/CED-3 protease activity. J Biol Chem. (1996) 271:28753–6. doi: 10.1074/jbc.271.46.28753
20. Amarante-Mendes GP, Finucane DM, Martin SJ, Cotter TG, Salvesen GS, Green DR. Anti-apoptotic oncogenes prevent caspase-dependent and independent commitment for cell death. Cell Death Differ. (1998) 5:298–306. doi: 10.1038/sj.cdd.4400354
21. Tait SW, Green DR. Mitochondrial regulation of cell death. Cold Spring Harb Perspect Biol. (2013) 5:a008706. doi: 10.1101/cshperspect.a008706
22. Wertz IE, Dixit VM. Regulation of death receptor signaling by the ubiquitin system. Cell Death Differ. (2010) 17:14–24. doi: 10.1038/cdd.2009.168
23. Guicciardi ME, Gores GJ. Life and death by death receptors. FASEB (2009) 23:1625–37. doi: 10.1096/fj.08-111005
24. Yu JW, Shi Y. FLIP and the death effector domain family. Oncogene (2008) 27:6216–27. doi: 10.1038/onc.2008.299
25. Salaun B, Romero P, Lebecque S. Toll-like receptors' two-edged sword: when immunity meets apoptosis. Eur J Immunol. (2007) 37:3311–8. doi: 10.1002/eji.200737744
26. Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, et al. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science (1999) 285:736–9. doi: 10.1126/science.285.5428.736
27. Aliprantis AO, Yang RB, Weiss DS, Godowski P, Zychlinsky A. The apoptotic signaling pathway activated by Toll-like receptor-2. EMBO J. (2000) 19:3325–36. doi: 10.1093/emboj/19.13.3325
28. Lopez M, Sly LM, Luu Y, Young D, Cooper H, Reiner NE. The 19-kDa Mycobacterium tuberculosis protein induces macrophage apoptosis through Toll-like receptor-2. J Immunol. (2003) 170:2409–16. doi: 10.4049/jimmunol.170.5.2409
29. Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell (2011) 43:449–63. doi: 10.1016/j.molcel.2011.06.011
30. Weber A, Kirejczyk Z, Besch R, Potthoff S, Leverkus M, Hacker G. Proapoptotic signalling through Toll-like receptor-3 involves TRIF-dependent activation of caspase-8 and is under the control of inhibitor of apoptosis proteins in melanoma cells. Cell Death Differ. (2010) 17:942–51. doi: 10.1038/cdd.2009.190
31. Salaun B, Coste I, Rissoan MC, Lebecque SJ, Renno T. TLR3 can directly trigger apoptosis in human cancer cells. J Immunol. (2006) 176:4894–901. doi: 10.4049/jimmunol.176.8.4894
32. Ruckdeschel K, Pfaffinger G, Haase R, Sing A, Weighardt H, Hacker G, et al. Signaling of apoptosis through TLRs critically involves toll/IL-1 receptor domain-containing adapter inducing IFN-beta, but not MyD88, in bacteria-infected murine macrophages. J Immunol. (2004) 173:3320–8. doi: 10.4049/jimmunol.173.5.3320
33. Haase R, Kirschning CJ, Sing A, Schrottner P, Fukase K, Kusumoto S, et al. A dominant role of Toll-like receptor 4 in the signaling of apoptosis in bacteria-faced macrophages. J Immunol. (2003) 171:4294–303. doi: 10.4049/jimmunol.171.8.4294
34. Kaiser WJ, Offermann MK. Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J Immunol. (2005) 174:4942–52. doi: 10.4049/jimmunol.174.8.4942
35. Harberts E, Fishelevich R, Liu J, Atamas SP, Gaspari AA. MyD88 mediates the decision to die by apoptosis or necroptosis after UV irradiation. Innate Immun. (2014) 20:529–39. doi: 10.1177/1753425913501706
36. Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol. (1988) 141:2629–34.
37. Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, et al. (1998). Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med. (1998) 188:919–30. doi: 10.1084/jem.188.5.919
38. Ray CA, Pickup DJ. The mode of death of pig kidney cells infected with cowpox virus is governed by the expression of the crmA gene. Virology (1996) 217:384–91. doi: 10.1006/viro.1996.0128
39. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. (2005) 1:112–9. doi: 10.1038/nchembio711
40. Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity (1996) 4:387–96. doi: 10.1016/S1074-7613(00)80252-6
41. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science (2009) 325:332–6. doi: 10.1126/science.1172308
42. Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell (2012) 150:339–50. doi: 10.1016/j.cell.2012.06.019
43. Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem. (2002) 277:9505–11. doi: 10.1074/jbc.M109488200
44. Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity (2013) 39:443–53. doi: 10.1016/j.immuni.2013.06.018
45. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell (2012) 148:213–27. doi: 10.1016/j.cell.2011.11.031
46. Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA. (2012) 109:5322–7. doi: 10.1073/pnas.1200012109
47. Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. (2013) 23:994–1006. doi: 10.1038/cr.2013.91
48. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. (2014) 16:55–65. doi: 10.1038/ncb2883
49. Chen X, Li W, Ren J, Huang D, He WT, Song Y, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. (2014) 24:105–21. doi: 10.1038/cr.2013.171
50. Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. (2014) 7:971–81. doi: 10.1016/j.celrep.2014.04.026
51. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell (2014) 54:133–46. doi: 10.1016/j.molcel.2014.03.003
52. Xia B, Fang S, Chen X, Hu H, Chen P, Wang H, et al. MLKL forms cation channels. Cell Res. (2016) 26:517–28. doi: 10.1038/cr.2016.26
53. Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, et al. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell (2017). 169:286.e16–300. doi: 10.1016/j.cell.2017.03.020
54. Weinlich R, Green DR. The two faces of receptor interacting protein kinase-1. Mol Cell (2014) 56:469–80. doi: 10.1016/j.molcel.2014.11.001
55. Grootjans S, Vanden Berghe T, Vandenabeele P. Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ. (2017) 24:1184–95. doi: 10.1038/cdd.2017.65
56. De Carvalho DD, Binato R, Pereira WO, Leroy JM, Colassanti MD, Proto-Siqueira R, et al. BCR-ABL-mediated upregulation of PRAME is responsible for knocking down TRAIL in CML patients. Oncogene (2011) 30:223–33. doi: 10.1038/onc.2010.409
57. Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe (2012) 11:290–7. doi: 10.1016/j.chom.2012.01.016
58. Schock SN, Chandra NV, Sun Y, Irie T, Kitagawa Y, Gotoh B, et al. Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway. Cell Death Differ. (2017) 24:615–25. doi: 10.1038/cdd.2016.153
59. Thapa RJ, Nogusa S, Chen P, Maki JL, Lerro A, Andrake M, et al. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci USA. (2013) 110:E3109–18. doi: 10.1073/pnas.1301218110
60. Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell (2014) 157:1189–202. doi: 10.1016/j.cell.2014.04.018
61. Orozco S, Yatim N, Werner MR, Tran H, Gunja SY, Tait SW, et al. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. (2014) 21:1511–21. doi: 10.1038/cdd.2014.76
62. Vanden Berghe T, Hassannia B, Vandenabeele P. An outline of necrosome triggers. Cell Mol Life Sci. (2016) 73:2137–52. doi: 10.1007/s00018-016-2189-y
63. Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell (2011) 43:432–48. doi: 10.1016/j.molcel.2011.06.006
64. Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, et al. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther. (2007) 6:1641–9. doi: 10.1158/1535-7163.MCT-06-0511
65. Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. (2012) 31:1062–79. doi: 10.1038/emboj.2011.497
66. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. (2009) 15:1170–8. doi: 10.1038/nm.2028
67. Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA. (2009) 106:20388–93. doi: 10.1073/pnas.0908698106
68. Adamczak SE, de Rivero Vaccari JP, Dale G, Brand FJ III, Nonner D, Bullock MR, et al. Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. J Cereb Blood Flow Metab. (2014) 34:621–9. doi: 10.1038/jcbfm.2013.236
69. Yang H, Ma Y, Chen G, Zhou H, Yamazaki T, Klein C, et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology (2016) 5:e1149673. doi: 10.1080/2162402X.2016.1149673
70. Weyd H, Abeler-Dorner L, Linke B, Mahr A, Jahndel V, Pfrang S, et al. Annexin A1 on the surface of early apoptotic cells suppresses CD8+ T cell immunity. PLoS ONE (2013) 8:e62449. doi: 10.1371/journal.pone.0062449
71. Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The adaptor ASC has extracellular and ‘prionoid' activities that propagate inflammation. Nat Immunol. (2014) 15:727–37. doi: 10.1038/ni.2913
72. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. (2007) 13:54–61. doi: 10.1038/nm1523
73. Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. (2009) 28:578–90. doi: 10.1038/emboj.2009.1
74. Kim H, Kim WJ, Jeon ST, Koh EM, Cha HS, Ahn KS, et al. Cyclophilin A may contribute to the inflammatory processes in rheumatoid arthritis through induction of matrix degrading enzymes and inflammatory cytokines from macrophages. Clin Immunol. (2005) 116:217–24. doi: 10.1016/j.clim.2005.05.004
75. Dear JW, Simpson KJ, Nicolai MP, Catterson JH, Street J, Huizinga T, et al. Cyclophilin A is a damage-associated molecular pattern molecule that mediates acetaminophen-induced liver injury. J Immunol. (2011) 187:3347–52. doi: 10.4049/jimmunol.1100165
76. Miles K, Clarke DJ, Lu W, Sibinska Z, Beaumont PE, Davidson DJ, et al. Dying and necrotic neutrophils are anti-inflammatory secondary to the release of alpha-defensins. J Immunol. (2009) 183:2122–32. doi: 10.4049/jimmunol.0804187
77. Calderwood SK, Gong J, Murshid A. Extracellular HSPs: the complicated roles of extracellular HSPs in Immunity. Front Immunol. (2016) 7:159. doi: 10.3389/fimmu.2016.00159
78. Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol. (2000) 12:1539–46. doi: 10.1093/intimm/12.11.1539
79. Pouwels SD, Zijlstra GJ, van der Toorn M, Hesse L, Gras R, Ten Hacken NH, et al. Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. Am J Physiol Lung Cell Mol Physiol. (2016) 310:L377–86. doi: 10.1152/ajplung.00174.2015
80. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. (2007) 13:1050–9. doi: 10.1038/nm1622
81. Bell CW, Jiang W, Reich CF III, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell physiol. (2006) 291:C1318–25. doi: 10.1152/ajpcell.00616.2005
82. Hou L, Yang Z, Wang Z, Zhang X, Zhao Y, Yang H, et al. NLRP3/ASC-mediated alveolar macrophage pyroptosis enhances HMGB1 secretion in acute lung injury induced by cardiopulmonary bypass. Lab Invest. (2018) 98:1052–64. doi: 10.1038/s41374-018-0073-0
83. Martinotti S, Patrone M, Ranzato E. Emerging roles for HMGB1 protein in immunity, inflammation, and cancer. Immunotargets Ther. (2015) 4:101–9. doi: 10.2147/ITT.S58064
84. Li H, Bo H, Wang J, Shao H, Huang S. Separation of supercoiled from open circular forms of plasmid DNA, and biological activity detection. Cytotechnology (2011) 63:7–12. doi: 10.1007/s10616-010-9322-9
85. Yang D, Postnikov YV, Li Y, Tewary P, de la Rosa G, Wei F, et al. High-mobility group nucleosome-binding protein 1 acts as an alarmin and is critical for lipopolysaccharide-induced immune responses. J Exp Med. (2012) 209:157–71. doi: 10.1084/jem.20101354
86. Murakami T, Hu Z, Tamura H, Nagaoka I. Release mechanism of high mobility group nucleosome binding domain 1 from lipopolysaccharide-stimulated macrophages. Mol Med Rep. (2016) 13:3115–20. doi: 10.3892/mmr.2016.4893
87. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity (2013) 38:209–23. doi: 10.1016/j.immuni.2013.02.003
88. Kim B, Lee Y, Kim E, Kwak A, Ryoo S, Bae SH, et al. The interleukin-1alpha precursor is biologically active and is likely a key alarmin in the IL-1 family of cytokines. Front Immunol. (2013) 4:391. doi: 10.3389/fimmu.2013.00391
89. England H, Summersgill HR, Edye ME, Rothwell NJ, Brough D. Release of interleukin-1alpha or interleukin-1beta depends on mechanism of cell death. J Biol Chem. (2014) 289:15942–50. doi: 10.1074/jbc.M114.557561
90. Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, et al. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity (2012) 36:388–400. doi: 10.1016/j.immuni.2012.01.018
91. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
92. Bonilla WV, Frohlich A, Senn K, Kallert S, Fernandez M, Johnson S, et al. The alarmin interleukin-33 drives protective antiviral CD8+ T cell responses. Science (2012) 335:984–9. doi: 10.1126/science.1215418
93. Vanden Berghe T, Kalai M, Denecker G, Meeus A, Saelens X, Vandenabeele P. Necrosis is associated with IL-6 production but apoptosis is not. Cell Signal. (2006) 18:328–35. doi: 10.1016/j.cellsig.2005.05.003
94. Coutant F, Perrin-Cocon L, Agaugue S, Delair T, Andre P, Lotteau V. Mature dendritic cell generation promoted by lysophosphatidylcholine. J Immunol. (2002) 169:1688–95. doi: 10.4049/jimmunol.169.4.1688
95. Rickard JA, O'Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell (2014) 157:1175–88. doi: 10.1016/j.cell.2014.04.019
96. Maeda A, Fadeel B. Mitochondria released by cells undergoing TNF-alpha-induced necroptosis act as danger signals. Cell Death Dis. (2014) 5:e1312. doi: 10.1038/cddis.2014.277
97. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464:104–7. doi: 10.1038/nature08780
98. Carlos D, Costa FR, Pereira CA, Rocha FA, Yaochite JN, Oliveira GG, et al. Mitochondrial DNA activates the NLRP3 inflammasome and predisposes to Type 1 diabetes in murine model. Front Immunol. (2017) 8:164. doi: 10.3389/fimmu.2017.00164
99. Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, et al. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci USA. (2014) 111:15514–9. doi: 10.1073/pnas.1414859111
100. Crouser ED, Shao G, Julian MW, Macre JE, Shadel GS, Tridandapani S, et al. Monocyte activation by necrotic cells is promoted by mitochondrial proteins and formyl peptide receptors. Crit Care Med. (2009) 37:2000–9. doi: 10.1097/CCM.0b013e3181a001ae
101. Noll F, Behnke J, Leiting S, Troidl K, Alves GT, Muller-Redetzky H, et al. Self-extracellular RNA acts in synergy with exogenous danger signals to promote inflammation. PLoS ONE (2017) 12:e0190002. doi: 10.1371/journal.pone.0190002
102. Choi JJ, Reich CF III, Pisetsky DS. Release of DNA from dead and dying lymphocyte and monocyte cell lines in vitro. Scand J Immunol. (2004) 60:159–66. doi: 10.1111/j.0300-9475.2004.01470.x
103. Matsumura K, Iwai H, Kato-Miyazawa M, Kirikae F, Zhao J, Yanagawa T, et al. Peroxiredoxin 1 contributes to host defenses against Mycobacterium tuberculosis. J Immunol. (2016) 197:3233–44. doi: 10.4049/jimmunol.1601010
104. Chen B, Miller AL, Rebelatto M, Brewah Y, Rowe DC, Clarke L, et al. S100A9 induced inflammatory responses are mediated by distinct damage associated molecular patterns (DAMP) receptors in vitro and in vivo. PLoS ONE (2015) 10:e0115828. doi: 10.1371/journal.pone.0115828
105. Lotfi R. Necrosis-Associated Factors (DAMPs) including S100A4 used to pulse Dendritic Cells (DCs) induce regulatory T cells. J Cell Sci Ther. (2013) 3:3–8. doi: 10.4172/2157-7013.1000134
106. Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol. (2008) 9:1179–88. doi: 10.1038/ni.1651
107. Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature (2016) 532:245–9. doi: 10.1038/nature17403
108. Kono H, Chen CJ, Ontiveros F, Rock KL. Uric acid promotes an acute inflammatory response to sterile cell death in mice. J Clin Invest. (2010) 120:1939–49. doi: 10.1172/JCI40124
109. Braga TT, Forni MF, Correa-Costa M, Ramos RN, Barbuto JA, Branco P, et al. Soluble uric acid activates the NLRP3 inflammasome. Sci Rep. (2017) 7:39884. doi: 10.1038/srep39884
110. Moriwaki K, Balaji S, McQuade T, Malhotra N, Kang J, Chan FK. The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity (2014) 41:567–78. doi: 10.1016/j.immuni.2014.09.016
111. Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O'Reilly L, et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity (2012) 36:215–27. doi: 10.1016/j.immuni.2012.01.012
112. Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D'Cruz AA, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. (2015) 6:6282. doi: 10.1038/ncomms7282
113. Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira R, da Silva C, et al. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8+ T cells. Science (2015) 350:328–34. doi: 10.1126/science.aad0395
114. Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, et al. Vaccination with necroptotic cancer cells induces efficient anti-tumor immunity. Cell Rep. (2016) 15:274–87. doi: 10.1016/j.celrep.2016.03.037
115. Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity (2011) 35:908–18. doi: 10.1016/j.immuni.2011.09.020
116. Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS, et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. (2015) 25:707–25. doi: 10.1038/cr.2015.56
117. Liu P, Xu B, Shen W, Zhu H, Wu W, Fu Y, et al. Dysregulation of TNFalpha-induced necroptotic signaling in chronic lymphocytic leukemia: suppression of CYLD gene by LEF1. Leukemia (2012) 26:1293–300. doi: 10.1038/leu.2011.357
118. He L, Peng K, Liu Y, Xiong J, Zhu FF. Low expression of mixed lineage kinase domain-like protein is associated with poor prognosis in ovarian cancer patients. Onco Targets Ther. (2013) 6:1539–43. doi: 10.2147/OTT.S52805
119. Su Z, Yang Z, Xie L, DeWitt JP, Chen Y. Cancer therapy in the necroptosis era. Cell Death Differ. (2016) 23:748–56. doi: 10.1038/cdd.2016.8
120. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell (2009) 137:1112–23. doi: 10.1016/j.cell.2009.05.037
121. Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH III, Ingram JP, et al. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe (2016) 20:13–24. doi: 10.1016/j.chom.2016.05.011
122. Hartmann BM, Albrecht RA, Zaslavsky E, Nudelman G, Pincas H, Marjanovic N, et al. Pandemic H1N1 influenza A viruses suppress immunogenic RIPK3-driven dendritic cell death. Nat Commun. (2017) 8:1931. doi: 10.1038/s41467-017-02035-9
123. Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, et al. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe (2015) 17:243–51. doi: 10.1016/j.chom.2015.01.003
124. Huang Z, Wu SQ, Liang Y, Zhou X, Chen W, Li L, et al. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe (2015) 17:229–42. doi: 10.1016/j.chom.2015.01.002
125. Wang X, Li Y, Liu S, Yu X, Li L, Shi C, et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci USA. (2014) 111:15438–43. doi: 10.1073/pnas.1412767111
126. Omoto S, Guo H, Talekar GR, Roback L, Kaiser WJ, Mocarski ES. Suppression of RIP3-dependent necroptosis by human cytomegalovirus. J Biol Chem. (2015) 290:11635–48. doi: 10.1074/jbc.M115.646042
127. Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, et al. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc Natl Acad Sci USA. (2014) 111:7391–6. doi: 10.1073/pnas.1403477111
128. Pearson JS, Giogha C, Muhlen S, Nachbur U, Pham CL, Zhang Y, et al. EspL is a bacterial cysteine protease effector that cleaves RHIM proteins to block necroptosis and inflammation. Nat Microbiol. (2017) 2:16258. doi: 10.1038/nmicrobiol.2016.258
129. Yeh WC, de la Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, et al. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science (1998) 279:1954–8. doi: 10.1126/science.279.5358.1954
130. Walsh CM, Wen BG, Chinnaiyan AM, O'Rourke K, Dixit VM, Hedrick SM. A role for FADD in T cell activation and development. Immunity (1998) 8:439–49. doi: 10.1016/S1074-7613(00)80549-X
131. Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity (1998) 9:267–76. doi: 10.1016/S1074-7613(00)80609-3
132. Hu ZQ, Zhao WH. Type 1 interferon-associated necroptosis: a novel mechanism for Salmonella enterica Typhimurium to induce macrophage death. Cell Mol Immunol. (2013) 10:10–2. doi: 10.1038/cmi.2012.54
133. Gaiha GD, McKim KJ, Woods M, Pertel T, Rohrbach J, Barteneva N, et al. Dysfunctional HIV-specific CD8+ T cell proliferation is associated with increased caspase-8 activity and mediated by necroptosis. Immunity (2014) 41:1001–12. doi: 10.1016/j.immuni.2014.12.011
134. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. (2017) 18:127–36. doi: 10.1038/nrm.2016.149
135. Kitur K, Parker D, Nieto P, Ahn DS, Cohen TS, Chung S, et al. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog. (2015) 11:e1004820. doi: 10.1371/journal.ppat.1004820
136. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. (2017) 277:61–75. doi: 10.1111/imr.12534
137. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. (2006) 8:1812–25. doi: 10.1111/j.1462-5822.2006.00751.x
138. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. (2009) 7:99–109. doi: 10.1038/nrmicro2070
139. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16:407–20. doi: 10.1038/nri.2016.58
140. Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe (2010) 8:471–83. doi: 10.1016/j.chom.2010.11.007
141. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science (2013) 341:1250–3. doi: 10.1126/science.1240988
142. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science (2013) 341:1246–9. doi: 10.1126/science.1240248
143. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514:187–92. doi: 10.1038/nature13683
144. Yang D, He Y, Munoz-Planillo R, Liu Q, Nunez G. Caspase-11 requires the Pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity (2015) 43:923–32. doi: 10.1016/j.immuni.2015.10.009
145. Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. (2012) 28:137–61. doi: 10.1146/annurev-cellbio-101011-155745
146. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526:666–71. doi: 10.1038/nature15541
147. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
148. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. (2015) 25:1285–98. doi: 10.1038/cr.2015.139
149. Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. (2017) 27:673–84. doi: 10.1016/j.tcb.2017.05.005
150. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA. (2016) 113:7858–63. doi: 10.1073/pnas.1607769113
151. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535:111–6. doi: 10.1038/nature18590
152. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535:153–8. doi: 10.1038/nature18629
153. Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. (2016) 35:1766–78. doi: 10.15252/embj.201694696
154. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. (2017) 8:14128. doi: 10.1038/ncomms14128
155. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature (2017) 547:99–103. doi: 10.1038/nature22393
156. Van Opdenbosch N, Van Gorp H, Verdonckt M, Saavedra PHV, de Vasconcelos NM, Gonçalves A, et al. Caspase-1 engagement and TLR-induced c-FLIP expression suppress ASC/Caspase-8-dependent apoptosis by inflammasome sensors NLRP1b and NLRC4. Cell Rep. (2017) 21:3427–44. doi: 10.1016/j.celrep.2017.11.088
157. Schneider KS, Gross CJ, Dreier RF, Saller BS, Mishra R, Gorka O, et al. The inflammasome drives GSDMD-independent secondary pyroptosis and IL-1 release in the absence of Caspase-1 protease activity. Cell Rep. (2017) 21:3846–59. doi: 10.1016/j.celrep.2017.12.018
158. Mascarenhas DPA, Cerqueira DM. Pereira SF, Castanheira FVS, Fernandes TD, Manin GZ, et al. Inhibition of caspase-1 or gasdermin-D enable caspase-8 activation in the Naip5/NLRC4/ASC inflammasome. PLoS Pathog. (2017) 13:e1006502. doi: 10.1371/journal.ppat.1006502
159. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. (2017) 17:151–64. doi: 10.1038/nri.2016.147
160. Jorgensen I, Lopez JP, Laufer SA, Miao EA. IL-1β, IL-18, and eicosanoids promote neutrophil recruitment to pore-induced intracellular traps following pyroptosis. Eur J Immunol. (2016) 46:2761–6. doi: 10.1002/eji.201646647
161. Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med. (2016) 213:2113–28. doi: 10.1084/jem.20151613
162. Wright JA, Bryant CE. The killer protein Gasdermin D. Cell Death Differ. (2016) 23:1897–8. doi: 10.1038/cdd.2016.100
163. Rauch I, Deets KA, Ji DX, von Moltke J, Tenthorey JL, Lee AY, et al. NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and−8. Immunity (2017) 46:649–59. doi: 10.1016/j.immuni.2017.03.016
164. Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, et al. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe (2014) 16:249–56. doi: 10.1016/j.chom.2014.07.002
165. Chen KW, Gross CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, et al. The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge. Cell Rep. (2014) 8:570–82. doi: 10.1016/j.celrep.2014.06.028
166. Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, et al. Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep. (2018) 22:2924–36. doi: 10.1016/j.celrep.2018.02.067
167. Monteleone M, Stow JL, Schroder K. Mechanisms of unconventional secretion of IL-1 family cytokines. Cytokine (2015) 74:213–8. doi: 10.1016/j.cyto.2015.03.022
168. Elliott EI, Sutterwala FS. Monocytes take their own path to IL-1beta. Immunity (2016) 44:713–5. doi: 10.1016/j.immuni.2016.03.015
169. Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science (2016) 352:1232–6. doi: 10.1126/science.aaf3036
170. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity (2018) 48:35.e6–44. doi: 10.1016/j.immuni.2017.11.013
171. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human monocytes engage an alternative inflammasome pathway. Immunity (2016) 44:833–46. doi: 10.1016/j.immuni.2016.01.012
172. Russo HM, Rathkey J, Boyd-Tressler A, Katsnelson MA, Abbott DW, Dubyak GR. Active Caspase-1 induces plasma membrane pores that precede pyroptotic lysis and are blocked by Lanthanides. J Immunol. (2016) 197:1353–67. doi: 10.4049/jimmunol.1600699
173. DiPeso L, Ji DX, Vance RE, Price JV. Cell death and cell lysis are separable events during pyroptosis. Cell Death Discov. (2017) 3:17070. doi: 10.1038/cddiscovery.2017.70
174. Lage SL, Amarante-Mendes GP, Bortoluci KR. Evaluation of pyroptosis in macrophages using cytosolic delivery of purified flagellin. Methods (2013) 61:110–6. doi: 10.1016/j.ymeth.2013.02.010
175. Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. (2015) 265:130–42. doi: 10.1111/imr.12287
176. Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 protects against bacteria that escape the vacuole. Science (2013) 339:975–8. doi: 10.1126/science.1230751
177. Sauer JD, Pereyre S, Archer KA, Burke TP, Hanson B, Lauer P, et al. Listeria monocytogenes engineered to activate the Nlrc4 inflammasome are severely attenuated and are poor inducers of protective immunity. Proc Natl Acad Sci USA. (2011) 108:12419–24. doi: 10.1073/pnas.1019041108
178. Warren SE, Duong H, Mao DP, Armstrong A, Rajan J, Miao EA, et al. Generation of a Listeria vaccine strain by enhanced caspase-1 activation. Eur J Immunol. (2011) 41:1934–40. doi: 10.1002/eji.201041214
179. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. (2010) 11:1136–42. doi: 10.1038/ni.1960
180. Mariathasan S, Weiss DS, Dixit VM, Monack DM. Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J Exp Med. (2005) 202:1043–9. doi: 10.1084/jem.20050977
181. Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, et al. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science (2014) 343:428–32. doi: 10.1126/science.1243640
182. Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature (2014) 505:509–14. doi: 10.1038/nature12940
183. Munoz-Arias I, Doitsh G, Yang Z, Sowinski S, Ruelas D, Greene WC. Blood-derived CD4 T cells naturally resist pyroptosis during abortive HIV-1 infection. Cell Host Microbe (2015) 18:463–70. doi: 10.1016/j.chom.2015.09.010
184. Ahmad F, Mishra N, Ahrenstorf G, Franklin BS, Latz E, Schmidt RE, et al. Evidence of inflammasome activation and formation of monocyte-derived ASC specks in HIV-1 positive patients. AIDS (2018) 32:299–307. doi: 10.1097/QAD.0000000000001693
185. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. (2005) 202:1691–701. doi: 10.1084/jem.20050915
186. Yatim N, Cullen S, Albert ML. Dying cells actively regulate adaptive immune responses. Nat Rev Immunol. (2017) 17:262–75. doi: 10.1038/nri.2017.9
187. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. (2017) 17:97–111. doi: 10.1038/nri.2016.107
188. Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med. (2010) 207:1807–17. doi: 10.1084/jem.20101157
189. Nagata S. Apoptotic DNA fragmentation. Exp Cell Res. (2000) 256:12–8. doi: 10.1006/excr.2000.4834
190. Trahtemberg U, Mevorach D. Apoptotic cells induced signaling for immune homeostasis in macrophages and dendritic cells. Front Immunol. (2017) 8:1356. doi: 10.3389/fimmu.2017.01356
191. Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity (2009) 31:84–98. doi: 10.1016/j.immuni.2009.05.007
192. Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity (2008) 29:21–32. doi: 10.1016/j.immuni.2008.05.013
193. Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity (2013) 38:729–41. doi: 10.1016/j.immuni.2013.03.003
194. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. (2014) 20:1301–9. doi: 10.1038/nm.3708
195. Vacchelli E, Ma Y, Baracco EE, Sistigu A, Enot DP, Pietrocola F, et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science (2015) 350:972–8. doi: 10.1126/science.aad0779
196. Torchinsky MB, Garaude J, Martin AP, Blander JM. Innate immune recognition of infected apoptotic cells directs T(H)17 cell differentiation. Nature (2009) 458:78–82. doi: 10.1038/nature07781
197. Campisi L, Barbet G, Ding Y, Esplugues E, Flavell RA, Blander JM. Apoptosis in response to microbial infection induces autoreactive TH17 cells. Nat Immunol. (2016) 17:1084–92. doi: 10.1038/ni.3512
198. Fond AM, Ravichandran KS. Clearance of dying cells by phagocytes: mechanisms and implications for disease pathogenesis. Adv Exp Med Biol. (2016) 930:25–49. doi: 10.1007/978-3-319-39406-0_2
199. Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. (2014) 14:166–80. doi: 10.1038/nri3607
200. El Mezayen R, El Gazzar M, Seeds MC, McCall CE, Dreskin SC, Nicolls MR. Endogenous signals released from necrotic cells augment inflammatory responses to bacterial endotoxin. Immunol Lett. (2007) 111:36–44. doi: 10.1016/j.imlet.2007.04.011
201. Nystrom S, Antoine DJ, Lundback P, Lock JG, Nita AF, Hogstrand K, et al. TLR activation regulates damage-associated molecular pattern isoforms released during pyroptosis. EMBO J. (2013) 32:86–99. doi: 10.1038/emboj.2012.328
202. Bonnet MC, Preukschat D, Welz PS, van Loo G, Ermolaeva MA, Bloch W, et al. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity (2011) 35:572–82. doi: 10.1016/j.immuni.2011.08.014
203. Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Hacker H, et al. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem. (2001) 276:31332–9. doi: 10.1074/jbc.M103217200
204. Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell (2010) 140:798–804. doi: 10.1016/j.cell.2010.02.015
205. Wang Q, Imamura R, Motani K, Kushiyama H, Nagata S, Suda T. Pyroptotic cells externalize eat-me and release find-me signals and are efficiently engulfed by macrophages. Int Immunol. (2013) 25:363–72. doi: 10.1093/intimm/dxs161
206. Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature (2003) 425:516–21. doi: 10.1038/nature01991
207. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature (2006) 440:237–41. doi: 10.1038/nature04516
208. Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. (2007) 13:851–6. doi: 10.1038/nm1603
209. Abdi J, Mutis T, Garssen J, Redegeld FA. Toll-like receptor (TLR)-1/2 triggering of multiple myeloma cells modulates their adhesion to bone marrow stromal cells and enhances bortezomib-induced apoptosis. PLoS ONE (2014) 9:e96608. doi: 10.1371/journal.pone.0096608
210. Eriksson M, Pena-Martinez P, Ramakrishnan R, Chapellier M, Hogberg C, Glowacki G, et al. Agonistic targeting of TLR1/TLR2 induces p38 MAPK-dependent apoptosis and NFkappaB-dependent differentiation of AML cells. Blood Adv. (2017) 1:2046–57. doi: 10.1182/bloodadvances.2017006148
211. Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. (2013) 288:31268–79. doi: 10.1074/jbc.M113.462341
212. Zhang Y, Bliska JB. Role of Toll-like receptor signaling in the apoptotic response of macrophages to Yersinia infection. Infect Immun. (2003) 71:1513–9. doi: 10.1128/IAI.71.3.1513-1519.2003
213. He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA. (2011) 108:20054–9. doi: 10.1073/pnas.1116302108
214. Sun R, Zhang Y, Lv Q, Liu B, Jin M, Zhang W, et al. Toll-like receptor 3 (TLR3) induces apoptosis via death receptors and mitochondria by up-regulating the transactivating p63 isoform alpha (TAP63alpha). J Biol Chem. (2011) 286:15918–28. doi: 10.1074/jbc.M110.178798
215. Huang Z, Zhou T, Sun X, Zheng Y, Cheng B, Li M, et al. Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through TLR4 activation. Cell Death Differ. (2018) 25:180–9. doi: 10.1038/cdd.2017.141
216. Ding HS, Yang J, Chen P, Yang J, Bo SQ, Ding JW, et al. The HMGB1-TLR4 axis contributes to myocardial ischemia/reperfusion injury via regulation of cardiomyocyte apoptosis. Gene (2013) 527:389–93. doi: 10.1016/j.gene.2013.05.041
217. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. (2008) 9:847–56. doi: 10.1038/ni.1631
218. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science (2008) 320:674–7. doi: 10.1126/science.1156995
219. Shio MT, Eisenbarth SC, Savaria M, Vinet AF, Bellemare MJ, Harder KW, et al. Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog. (2009) 5:e1000559. doi: 10.1371/annotation/abca067d-b82b-4de6-93c5-0fcc38e3df05
220. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature (2010) 464:1357–61. doi: 10.1038/nature08938
221. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature (2006) 440:228–32. doi: 10.1038/nature04515
222. Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E, et al. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE (2009) 4:e7446. doi: 10.1371/journal.pone.0007446
223. Gurcel L, Abrami L, Girardin S, Tschopp J, van der Goot FG. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell (2006) 126:1135–45. doi: 10.1016/j.cell.2006.07.033
224. Harder J, Franchi L, Munoz-Planillo R, Park JH, Reimer T, Nunez G. Activation of the Nlrp3 inflammasome by Streptococcus pyogenes requires streptolysin O and NF-kappa B activation but proceeds independently of TLR signaling and P2X7 receptor. J Immunol. (2009) 183:5823–9. doi: 10.4049/jimmunol.0900444
225. Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature (2006) 440:233–6. doi: 10.1038/nature04517
226. Franchi L, Eigenbrod T, Munoz-Planillo R, Ozkurede U, Kim YG, Arindam C, et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J Immunol. (2014) 193:4214–22. doi: 10.4049/jimmunol.1400582
227. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. (2011) 12:408–15. doi: 10.1038/ni.2022
228. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature (2011) 477:592–5. doi: 10.1038/nature10394
229. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature (2011) 477:596–600. doi: 10.1038/nature10510
230. Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol. (2006) 7:576–82. doi: 10.1038/ni1346
231. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol. (2006) 7:569–75. doi: 10.1038/ni1344
232. Rauch I, Tenthorey JL, Nichols RD, Al Moussawi K, Kang JJ, Kang C, et al. NAIP proteins are required for cytosolic detection of specific bacterial ligands in vivo. J Exp Med. (2016) 213:657–65. doi: 10.1084/jem.20151809
233. Zhao Y, Shi J, Shi X, Wang Y, Wang F, Shao F. Genetic functions of the NAIP family of inflammasome receptors for bacterial ligands in mice. J Exp Med. (2016) 213:647–56. doi: 10.1084/jem.20160006
234. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458:514–8. doi: 10.1038/nature07725
235. Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. (2006) 38:240–4. doi: 10.1038/ng1724
236. Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell (2007) 25:713–24. doi: 10.1016/j.molcel.2007.01.032
237. Xu H, Yang J, Gao W, Li L, Li P, Zhang L, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature (2014) 513:237–41. doi: 10.1038/nature13449
238. Salvestrini V, Orecchioni S, Talarico G, Reggiani F, Mazzetti C, Bertolini F, et al. Extracellular ATP induces apoptosis through P2X7R activation in acute myeloid leukemia cells but not in normal hematopoietic stem cells. Oncotarget (2017) 8:5895–908. doi: 10.18632/oncotarget.13927
239. Placido R, Auricchio G, Falzoni S, Battistini L, Colizzi V, Brunetti E, et al. P2X(7) purinergic receptors and extracellular ATP mediate apoptosis of human monocytes/macrophages infected with Mycobacterium tuberculosis reducing the intracellular bacterial viability. Cell Immunol. (2006) 244:10–8. doi: 10.1016/j.cellimm.2007.02.001
240. Ishibashi O, Ali MM, Luo SS, Ohba T, Katabuchi H, Takeshita T, et al. Short RNA duplexes elicit RIG-I-mediated apoptosis in a cell type- and length-dependent manner. Sci Signal. (2011) 4:ra74. doi: 10.1126/scisignal.2001614
Keywords: PRR, pathogen recognition receptor, apoptosis, necroptosis, pyroptosis, inflammation
Citation: Amarante-Mendes GP, Adjemian S, Branco LM, Zanetti LC, Weinlich R and Bortoluci KR (2018) Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 9:2379. doi: 10.3389/fimmu.2018.02379
Received: 02 August 2018; Accepted: 25 September 2018;
Published: 16 October 2018.
Edited by:
Leticia A. Carneiro, Universidade Federal do Rio de Janeiro, BrazilReviewed by:
Fabianno Ferreira Dutra, Universidade Federal do Estado do Rio de Janeiro, BrazilMarco Di Gioia, Boston Children's Hospital, Harvard University, United States
Copyright © 2018 Amarante-Mendes, Adjemian, Branco, Zanetti, Weinlich and Bortoluci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gustavo P. Amarante-Mendes, gpam@usp.br
Ricardo Weinlich, ricardo.weinlich@einstein.br
Karina R. Bortoluci, kbortoluci@gmail.com