 Valeria Caneparo
Valeria Caneparo Santo Landolfo
Santo Landolfo Marisa Gariglio
Marisa Gariglio Marco De Andrea
Marco De Andrea- 1Viral Pathogenesis Unit, Department of Public Health and Pediatric Sciences, Turin Medical School, Turin, Italy
- 2Virology Unit, Interdisciplinary Research Center of Autoimmune Diseases (IRCAD), Department of Translational Medicine, Novara Medical School, Novara, Italy
- 3Intrinsic Immunity Unit, CAAD – Center for Translational Research on Autoimmune and Allergic Disease, University of Piemonte Orientale, Novara, Italy
Absent in melanoma 2 (AIM2)-like receptors (ALRs) are a newly characterized class of pathogen recognition receptors (PRRs) involved in cytosolic and nuclear pathogen DNA recognition. In recent years, two ALR family members, the interferon (IFN)-inducible protein 16 (IFI16) and AIM2, have been linked to the pathogenesis of various autoimmune diseases, among which systemic lupus erythematosus (SLE) has recently gained increasing attention. SLE patients are indeed often characterized by constitutively high serum IFN levels and increased expression of IFN-stimulated genes due to an abnormal response to pathogens and/or incorrect self-DNA recognition process. Consistently, we and others have shown that IFI16 is overexpressed in a wide range of autoimmune diseases where it triggers production of specific autoantibodies. In addition, evidence from mouse models supports a model whereby ALRs are required for IFN-mediated host response to both exogenous and endogenous DNA. Following interaction with cytoplasmic or nuclear nucleic acids, ALRs can form a functional inflammasome through association with the adaptor ASC [apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD)] and with procaspase-1. Importantly, inflammasome-mediated upregulation of IL-1β and IL-18 production positively correlates with SLE disease severity. Therefore, targeting ALR sensors and their downstream pathways represents a promising alternative therapeutic approach for SLE and other systemic autoimmune diseases.
Introduction
Inflammation is an innate immune response largely mediated by phagocytic cells whose goal is to protect the body from invading bacteria and viruses (1, 2). Pattern recognition receptors (PRRs) constitute a large family of molecules expressed on the cell surface and in the cytoplasm of various cell types, such as macrophages and antigen presenting cells (APC), able to interact with evolutionarily conserved pathogenic structures [i.e., pathogen-associated molecular patterns (PAMPs)], thus giving rise to multimeric protein complexes termed inflammasomes, which are then responsible for mediating a caspase-1-dependent inflammatory response (3–6). These so-called “canonical inflammasomes,” which can be triggered by a wide variety of ligands, consist of nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) and absent in melanoma 2 (AIM2)-like receptors (ALRs) (7–10). More recently, “non-canonical inflammasomes,” containing caspase-11 in mice and caspase-4/5 in humans, have also been described (11, 12).
Chronic inflammatory responses, which could last for weeks or even years, are characterized by episodes of tissue injury and healing resulting in severe tissue damage, which can eventually lead to the development of autoinflammatory/autoimmune diseases such as systemic lupus erythematosus (SLE) (13–15). This latter is an autoimmune disease characterized by a wide range of clinical and serological manifestations accompanied by a polyclonal autoimmune response against various nuclear autoantigens (16). Although genetic and environmental factors such as infections are known to be involved in the pathogenesis of SLE, the clear etiology of this disease still remains to be fully established (17).
Despite this gap in knowledge, it is now clear that ALRs, especially the IFN-inducible protein 16 (IFI16, Figure 1), along with other inflammasome-induced inflammatory responses, contribute to the development of SLE. In this review, we will summarize recent advances on the role of the inflammasome and ALRs in SLE, which could ultimately provide the rationale for the design and development of novel therapeutic tools for the treatment of patients affected by SLE or other systemic autoimmune diseases.
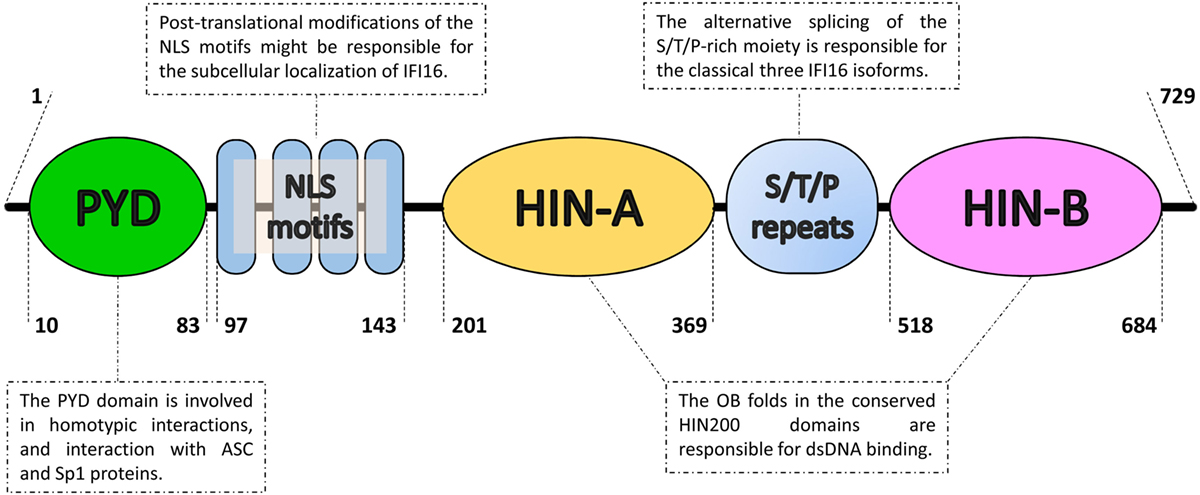
Figure 1. Domain organization of the IFN-inducible protein 16 (IFI16). From the N- to the C-terminal (left to right), IFI16 comprises a pyrin domain (PYD) involved in protein–protein interactions, a linker region containing four nuclear localization signal motifs (NLS) and two hematopoietic interferon-inducible nuclear protein with 200-amino-acid repeats (HIN200) domains, which is an hallmark of the absent in melanoma 2-like receptors/PYHIN proteins. The HIN200 domains include two tandem b-barrels, known as oligonucleotide–oligosaccharide-binding (OB) fold, which allow DNA docking in a non-sequence-specific manner. They are separated by serine/threonine/proline-rich (S/T/P) repeats, which are regulated by alternative mRNA splicing. The numbers represent the amino acid positions based on NCBI Reference Sequence NP_005522.2.
The Inflammasome
The canonical inflammasome is a multimodular complex that, upon induced oligomerization, stimulates the activation of caspase-1, an enzyme primarily responsible for the release of the pro-inflammatory cytokines IL-1β and IL-18 (6). Strongly associated with the activation of the inflammasome, pyroptosis is a caspase-1-dependent type of inflammatory cell death mainly seen during intracellular infections (18). Inflammasomes specific for intracellular PAMPs involve different classes of cytoplasmic PRRs. Classically, the NLR, such as NLRP3, and the retinoic acid inducible gene I (RIG-I)-like receptor (RLR) families (Figure 2).
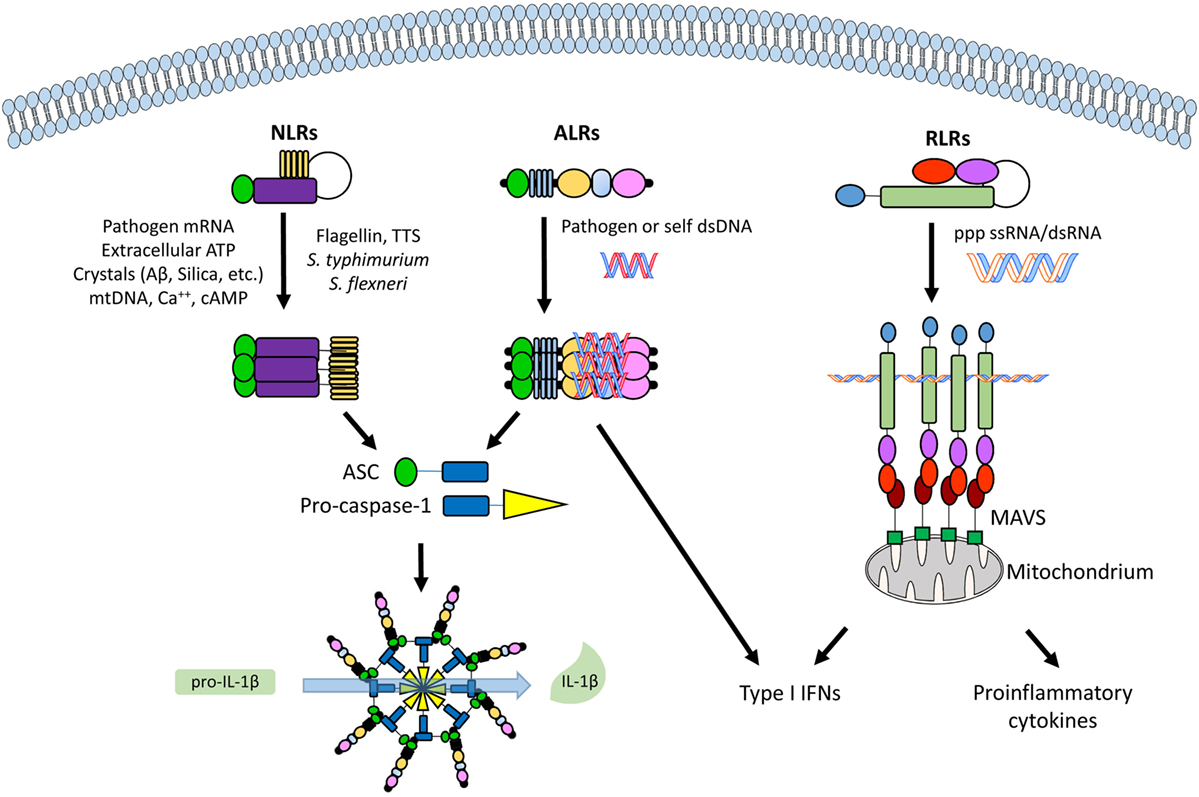
Figure 2. An overview of the different classes of cytoplasmic and nuclear pathogen recognition receptors (PRRs) and their involvement with inflammasome activation. From left to right, a nucleotide-binding oligomerization domain-like receptor (NLR, e.g., NLRP3), an absent in melanoma 2 (AIM2)-like receptor (ALR, e.g., IFI16), and a retinoic acid inducible gene I (RIG-I)-like receptor (RLR, e.g., RIG-I), with their commonest ligands. See text for details. Abbreviations: ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD); MAVS, mitochondrial antiviral signaling protein.
NLRP3 holds a C-terminal leucine-rich repeat domain, a central nucleotide-binding and oligomerization domain (NOD or NACHT), and an N-terminal pyrin domain (PYD). The NLR-associated PYD interacts with the PYD of the adaptor apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD) (ASC). ASC is then able to engage caspase-1 through its CARD domain causing the oligomerization of several caspase-1 molecules that, in turn, cleave and activate each other (8). RIG-I is made of two N-terminal CARDs, a central RNA helicase domain and a C-terminal regulatory domain (CTD). As for ASC, the RIG-I CARD is a sticky domain responsible for recruiting adaptor proteins and triggering downstream pathways (19). Whereas the RNA helicase domain contains a conserved Asp–Glu–Ala-Asp motif, also known as DEAD box, and exerts ATPase activity, the CTD is responsible for binding dsRNA PAMPs (20). Following dsRNA binding and associated conformational changes, RIG-I interacts with mitochondrial outer membrane proteins called mitochondrial antiviral signaling (MAVS) through CARD–CARD interactions (21). Depending on the adaptors involved, RIG-I–MAVS interaction then results in either type I IFN (IFN-I) or pro-inflammatory cytokines production (22).
Interestingly, recent studies have shown that there also exist non-canonical inflammasomes, which, through recruitment of caspase-4/5 in human or caspase-11 in mouse, can induce caspase-1-dependent maturation and secretion of IL-1β and IL-18 (23–25). In particular, non-canonical inflammasomes appear to promote pyroptosis in a TLR4- and caspase-1-independent fashion in response to cytoplasmic Gram-negative bacteria infection (26). Although the innate immune response mediated by caspase-4/5 resembles, at least in term of outcomes, that driven by caspase-1, studies on macrophage-mediated inflammatory responses have revealed that they are indeed two quite different processes (12, 27, 28). In human cells, in fact, the CARD motif allows pro-caspase-4/5 to directly interact with lipopolysaccharide (LPS) through the lipid A moiety leading to pro-caspase-4/5 oligomerization and induction of pyroptosis coupled with secretion of IL-1β and IL-18. Adding complexity to this scenario, recent evidence has shown that murine caspase-11 activation triggers an NLRP3–ASC–caspase-1-dependent signaling pathway, also known as “non-canonical NLRP3 inflammasome activation pathway,” which is different from the aforementioned “canonical NLRP3 inflammasome activation pathway” (29). However, even though it appears that caspase-4/5 and -11 can directly detect intracellular LPS derived from Gram-negative bacteria (24, 30), the exact mechanism of the non-canonical inflammasome activation is not totally understood.
Recently, a new family of inflammasome-associated PRRs has been described, including AIM2 and IFI16, grouped as ALRs. ALRs can assemble inflammasomes that respond to DNA molecules in both the cytosol and nucleus (31–33). AIM2 and IFI16 display an N-terminal PYD and one (AIM2) or two (IFI16, Figure 1) phylogenetically conserved hematopoietic interferon (IFN)-inducible nuclear protein with a 200-amino-acid repeat (HIN200) domains at the C-terminus, thus the other name PYHIN (or PYHIN200) previously given to these proteins. Interestingly, the HIN200 domain, which consists of two oligonucleotide/oligosaccharide-binding (OB) folds (34), appears to be the major DNA recognition site (35, 36). However, due to the lack of a dedicated ATP-dependent oligomerization domain, it appears that ALRs require a longer stretch of double-stranded DNA (dsDNA) compared with that required by NLRs to bind effectively and promote oligomerization (37, 38). Notably, since DNA is a common genetic material, pathological stimulation of these nucleic acid-recognizing inflammasomes by self-DNA can lead to autoinflammatory/autoimmune diseases as well (Figure 2). In this regard, aberrant immune responses involving ALRs have long been involved in the pathogenesis of SLE, Sjogren’s syndrome (SjS), psoriasis, and systemic sclerosis (SSc) (39–45).
Inflammasome and Autoimmunity
Although adaptive and innate responses are often opposite to each other in the immunological spectrum, they are essentially integrated in a complex system (i.e., the human body) as innate immune dysregulation (i.e., the classical driver of autoinflammatory diseases) induces autoreactive T and B cell responses (i.e., autoimmunity) (13, 46). Indeed, classical autoinflammatory diseases, such as inflammatory bowel disease (IBD), are also characterized by the presence of autoantibodies, whereas classical autoimmune conditions, such as SLE, can also display organ-specific inflammation, as in the case of lupus nephritis (LN) (47, 48).
An additional feature encompassing the full inflammatory spectrum is inflammasome activation, which is usually essential for host defense against microbes. However, recent studies have also found this activation to be responsible for or simply be associated with the pathogenesis of several diseases featuring autoinflammatory/autoimmune traits such as type 1 and type 2 diabetes, IBD, multiple sclerosis (MS), rheumatoid arthritis (RA), and SLE (49–55).
Genetic polymorphisms (SNPs) associated with autoimmune diseases have been identified in components of both NLR and ALR inflammasomes, including NLRP1, NLRP3, CARD8, IFI16, and AIM2 (52, 56–61). Several studies, most of which related to ethnicity, have highlighted an association between SNPs in inflammasome end products and autoimmune diseases such as SLE, RA, and MS (62–64). Furthermore, inflammasomes have been directly involved in autoimmunity. For example, NLRP1 is overexpressed in T and Langerhans cells in the leading edge of vitiligo skin, leading to increased IL-1β production and activation of the Th17 axis (65). Furthermore, NLRP3 expression and NLRP3-mediated IL-1β secretion are increased in RA patients (66), and NLRP3 is involved in the pathogenesis of experimental autoimmune encephalomyelitis (51). Moreover, APC with an activated NLRP3 inflammasome can trigger CD4 T cell-mediated upregulation of the chemokine receptor CCR2, which is elevated in the peripheral blood of MS patients during relapse (67). With regard to ALR family members, AIM2 is directly activated by cytoplasmic DNA (68), and a strong correlation between AIM2 overexpression and disease severity has been described in both SLE patients and mouse models (61, 69). Finally, SLE is characterized by AIM2 inflammasome-mediated production of IL-1β, triggered by accumulation of cytosolic self-DNA and IFI16-induced IFN-I release (40).
Systemic lupus erythematosus is a systemic autoimmune disease characterized by a polyclonal autoimmune response against various nuclear autoantigens (16). Although genetic and environmental factors, such as infections, have been linked to the pathogenesis of SLE, the exact etiology of this disease is still unknown (17). SLE is characterized by hyperactive autoreactive immune cells and production of many autoantibodies, immune complex (IC) formation, organ inflammation and damage. More than 200 different autoantibodies including those against single-stranded DNA and dsDNA, Ro/La antigens, and ribonuclear protein have been described in lupus patients (70, 71). Among these, the so-called antinuclear antibodies (ANAs) and anti-dsDNA antibodies, which seem to play an important role in the pathogenesis of LN, represent valuable diagnostic and prognostic markers of disease (72, 73).
Along with elevated production of autoantibodies, 50–75% of SLE adults and up to 90% of SLE children display increased IFN-I production and enhanced expression of IFN-inducible genes, which is therefore regarded as a gene expression signature of SLE (74). Notably, a few studies have shown that patients with enhanced IFN-I signature can be considered a distinct subset of SLE patients. In this context, an association between IFN signature and some clinical manifestations, such as nephritis and CNS disease, has been reported (75).
Recent studies have defined the role of autologous dsDNA in SLE pathogenesis [reviewed in Ref. (72)]. Briefly, in physiological conditions, dsDNA is localized in the nucleus and mitochondria; however, once it relocalizes into the cytoplasm and endosomes, it is rapidly degraded by DNases. In SLE patients, impaired dsDNA degradation activity coupled with defective clearance of both apoptotic cell bodies and neutrophil extracellular traps (NETs) results in self-dsDNA accumulation (76, 77). In the meantime, self-dsDNA released by apoptotic cells in the germinal center is processed by follicular dendritic cells as non-self-antigen and presented to autoreactive B cells, which as a result will survive and expand instead of being eliminated (78). Afterward, self-dsDNA together with autoantibodies triggers the formation of ICs that in turn will mediate tissue damage, stimulate pro-inflammatory cytokine production and an array of IFN-inducible genes (i.e., IFN signature). Noteworthy, self-dsDNA is mainly sensed by plasmacytoid dendritic cells (pDCs) by means of different DNA sensors, which ultimately lead to elevated IFN-I production and inflammasome activation (70).
Type I IFNs are endowed of several immune functions ranging from dendritic cell differentiation and maturation to T cells activation and induction of antibody production by B cells. IFN-I pleiotropic activities underscore the critical function of these molecules in the pathogenesis of autoimmune diseases, in particular SLE (70, 75). In parallel, inflammasome activation leads to the release of inflammatory cytokines including IL-1β and IL-18, which contributes to the maintenance of the inflammatory state followed by cell death.
However, the association between SLE and IL-1β production is highly debated. Animal models of SLE (MRL/lpr mice) have shown that IL-1β gene expression, and protein secretion is increased in the glomerular macrophages and mesangial cells of LN (79), whereas polymorphisms studies on SLE patients have led to conflicting results (80).
Altogether, these observations stress the relevant role of IFN-I alongside the other inflammatory cytokines in fine-tuning both the innate and adaptive immune responses. One can therefore easily understand how slight perturbations of the signaling pathways can lead to the dysregulation of the immune response that inevitably brings to the development of the autoimmune response.
Role of Autologous dsDNA in SLE
The major source of autologous dsDNA, which, as mentioned earlier, plays a pivotal role in SLE pathogenesis, is represented by cells dying by necrosis, apoptosis or NETosis, with the latter being a type of cell death mediated by NETs, extrusions of intracellular material to the surrounding extracellular medium to concentrate antibacterial substances and entrap invading microorganisms (81, 82). Intriguingly, also pyroptosis, that is the type of cell death induced by the inflammasome in response to both infectious and non-infectious stimuli, has been linked to SLE initiation (83).
Apoptosis, also known as programmed cell death, is an essential mechanism of tissue homeostasis during development and aging, characterized by cell shrinkage, cytoskeleton remodeling, chromatin condensation, nuclear breakup, plasma membrane blebbing and formation of typical apoptotic bodies (84). Under normal physiologic conditions, apoptotic cells directly undergo phagocytosis by specialized cells (i.e., professional phagocytes) and are degraded within the lysosomes with no signs of inflammation or immune response. In physiological conditions, cellular membranes are well preserved and readily cleared by engulfing phagocytes (85). Unless properly cleared, the apoptotic cells undergo secondary necrosis characterized by cell membrane leakage with consequent release of intracellular contents, including autologous dsDNA (86). Notably, release of intracellular material, which ultimately contributes to the development of autoimmune diseases, can also be triggered by primary necrosis due to exogenous factors, as demonstrated both in animal models and human infections (87, 88).
NETosis, a type of cell death first associated with neutrophils, causes the extrusion of nuclear DNA, histones and granular antimicrobial proteins entrapped leading to formation of NETs (81, 89). Yet, mounting evidence has shown that other cell types, including eosinophils and mast cells, can undergo cell death through a similar mechanism. Therefore, NETosis appears not be limited to neutrophils and should therefore be regarded as a new type of cell death that generally causes the release of extracellular traps (90). Physiologically, monocyte-derived phagocytes clear NETs efficiently thanks to C1q- and DNase I-mediated extracellular preprocessing of NETs. After ingestion by phagocytes, NETs are degraded in the lysosomes. Remarkably, this entire process is immunologically silent since the uptake of NETs by macrophages does not seem to stimulate pro-inflammatory cytokine secretion (91). On the other hand, impaired clearance of NETs by phagocytes can lead to the accumulation of several autoantigens including self-dsDNA (92), thereby increasing the chance of anti-dsDNA antibody formation, although a study on an animal model of SLE showed a protective role of NETs (93).
A particular type of NETosis, mitochondrial NETosis, causes the release of mitochondrial DNA (mtDNA) from neutrophils following the mitochondrial production of ROS. Since mitochondria share several features with bacteria, including a circular genome carrying unmethylated CpG dinucleotide repeats, mtDNA is similarly immunogenic and may promote inflammation through surface and endoplasmic TLR9 binding. Moreover, IL-1β production can also be driven by cytosolic release of mtDNA, dominantly acting on NLRP3/AIM2 inflammasomes (94). Interestingly, NETs from low-density granulocyte of SLE patients are highly enriched in mtDNA compared with NETs from healthy controls neutrophils (95), whereas abnormal extrusion of oxidized mtDNA from SLE patient neutrophils may triggers a pathogenic interferogenic response (96). Finally, mtDNA and autoantibodies against it are present in elevate levels in SLE and in particular in LN, where levels correlate with activity index better than anti-dsDNA (97).
Altogether, these findings indicate that cell death-originating self-dsDNA plays a crucial role in SLE pathogenesis.
Environmental Factors Triggering IFN-I Production and Inflammasome Activation in SLE
We have beforehand described that DNA from dying cells, as well as DNA from microbial pathogens, is strong immune stimulants that can accumulate in the cytosol and activate the production of various immune system modulators, including IFN-I. This pathway is critically dependent on a protein known as stimulator of interferon genes (STING) (98), which indirectly responds to DNA through the cyclic dinucleotide 2′,3′-cGAMP, produced upon the stimulation of the enzyme cyclic GMP-AMP synthase (cGAS) (99). In turn, the 2′,3′-cGAMP-related activation of STING induces a conformational change which is thought to mediate the phosphorylation and activation of interferon regulatory factor 3 (IRF3), a transcription factor for various gene targets, including but not limited to IFN-I (100).
It is becoming increasingly clear how several environmental factors that can promote IFN-I production are also able to induce an SLE syndrome as well as cause a flare of this disease. One of these agents is represented by ultraviolet B (UVB) light, which has been shown to trigger SLE flares and induce severe systemic manifestations including cutaneous reactions (101). Interestingly, all UVB light-induced exacerbations are associated with enhanced levels of IFN-I and -III along with pro-inflammatory cytokines (102, 103). In this regard, UVB light can promote redistribution of nuclear antigens on the cell surface and keratinocyte apoptosis (104). Furthermore, additional inflammatory cells, recruited by type III IFN into the skin, are likely responsible for priming activated pDCs to produce higher levels of IFN-I. Consistently, UV irradiation of keratinocytes has been shown to activate the STING/IRF regulatory axis in response to cytosolic DNA due to the loss of the STING negative regulator Unc51-like kinase 1 (105).
Systemic lupus erythematosus onset along with disease flare is also frequently associated with infections. Although many viruses and bacteria have been implicated in SLE pathogenesis (88, 106, 107), no specific etiologic pathogen has thus far been identified. Inflammation, as part of the innate immune response, is triggered when PAMPs are recognized by PRRs, which can be either associated with the cell membrane or located within the cell in the cytosol or nucleus. There is a growing number of identified PRRs, including toll-like receptors (TLRs) and various intracellular nucleic acid receptors. The signaling pathway leading to IFN-I production or inflammasome activation strictly relies on the PRR repertoire of the responding cell type and the subcellular localization of the immunostimulatory nucleic acid. TLR3, TLR7/8, and TLR9, present in immune cells (i.e., pDCs and monocytes), sense dsRNA, ssRNA, and DNA containing CpG motifs (108, 109). Another group of PRRs (i.e., the RLRs) include the cytosolic RNA receptor RIG-I and the melanoma differentiation factor 5, and are responsible of detecting dsRNA and ssRNA molecules in the cytoplasm of cells infected with RNA viruses (20, 110–112). In addition, several DNA sensors located in both the cytosol and the nucleus have been described. These include cGAS (113), DNA-dependent activator of IFN-regulatory factors (DAI) (114), AIM2 (115), IFI16 (116, 117), NLRs (118), and DEAD/H-box helicase 41 (DDX41) (119). Binding of these DNA sensors to their ligands activates signaling pathways, including TLR9-, STING-, and inflammasome-dependent pathways, which not only induce production of IFN-I but also promote inflammatory gene expression and inflammasome-associated cell death (i.e., pyroptosis). In physiological conditions, these intracellular sensors and related pathways are tightly regulated to impede the development of autoimmunity (120), which would otherwise take place due to uncontrolled recognition of self-nucleic acids (121, 122).
Upon PAMP recognition, the intracellular receptors assemble cytoplasmic platforms known as myddosomes and inflammasomes, which are supramolecular organizing centers regulating the inflammatory and immunoregulatory response following microbial detection. Specifically, TLRs initiate a toll/interleukin-1 receptor domain-containing adapter protein (TIRAP)-dependent assembly of the myddosome, which consists of the adaptor MYD88 and several serine/threonine kinases of the IL-1 receptor-associated kinase family (123). As stated previously, the canonical inflammasome contains a DNA sensor protein, the adaptor protein ASC and procaspase-1. Upon inflammasome assembling, activation of caspase-1 converts the immature IL-1β and IL-18 into mature secreted forms (124). Importantly, different NLR family members, such as NLRC4, NLRP1, and NLRP3 and the two ALR family members AIM2 and IFI16 have been shown to be differentially stimulated in a ligand-specific fashion.
Recently, it has been demonstrated that the canonical inflammasome pathway can be by-passed by the non-canonical one, which as stated previously consists of a complex formed by procaspase-11 and bacterial LPS activated in mouse macrophages. Consistently, caspase-4 and caspase-5, the human counterpart of mouse caspase-11, can interact directly with intracellular LPS and activate the non-canonical inflammasome in human myeloid cells (12, 23).
Two important features distinguish myddosomes from inflammasomes: (1) inflammasomes do not trigger gene activation at the transcriptional level, but rather induce inflammation by promoting the release of preexisting immature cytokines; (2) inflammasomes activating PRRs are localized in the host cytosol, which is rarely attacked by non-pathogenic bacteria. Therefore, inflammasomes are generally assembled when intracellular PRRs interact with pathogenic bacteria in the cytosol. By contrast, TLRs, which are localized on the cell surface, cannot distinguish whether PAMPs originated from pathogenic or non-pathogenic microorganisms.
Thus, taken together, these findings suggest a scenario where the redundancy of PAMPs sensing immune receptors may easily lead to dysregulation of the immune response when not regulated properly.
AIM2-Like Receptors: Inflammasome Activators and IFN-I Production Regulators
The PYHIN (or PYHIN200) family encodes evolutionary related human (i.e., IFI16, IFIX, MNDA, and AIM2) and murine (i.e., Ifi202a, Ifi202b, Ifi203, Ifi204, Ifi205/D3, and Ifi206) proteins (116–118). Increasing evidence has shown that these proteins may act as regulators of a wide range of cell functions, such as differentiation, proliferation, senescence, apoptosis, and inflammasome assembly (117, 125–130). Recently, two members of the human family, IFI16 and AIM2, have been implicated in the recognition of pathogen DNA and classified into the ALR group, still maintaining their peculiarity. In normal conditions, expression of the nuclear phosphoprotein IFI16 is limited to vascular endothelial cells, keratinocytes, and hematopoietic cells (131). Following activation by pathogen DNA, IFI16 translocates into the cytoplasm, triggers type I IFN production, cytokines, and eventually cell death (Figure 3). By contrast, AIM2, upon binding DNA in the cytosol, stimulates inflammasome activation in the absence of type I IFN production.
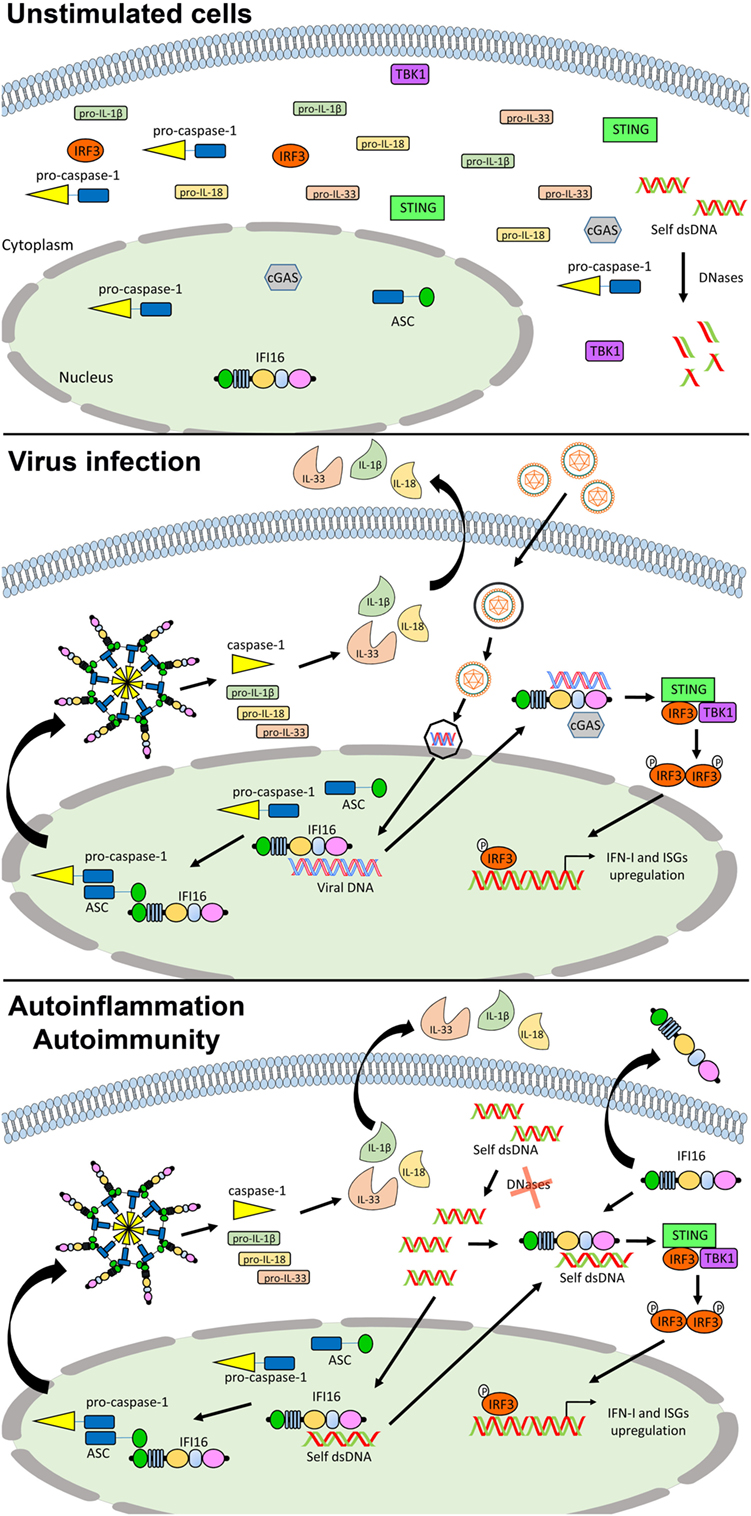
Figure 3. Role of IFN-inducible protein 16 (IFI16) as inflammasome regulator in viral infections and autoimmunity. In unstimulated cells, IFI16 is mainly nuclear (upper panel). Following viral DNA recognition and binding, IFI16 can induce the activation of the canonical inflammasome through the recruitment of ASC and pro-caspase 1, and the production of type I IFN (IFN-I) through the STING–IRF axis (middle panel). In the course of autoimmune (e.g., systemic lupus erythematosus) and autoinflammatory conditions, following the recognition of self-DNA, IFI16 might be responsible for the production of pro-inflammatory cytokines and IFN-I through the same pathways. Moreover, its aberrant expression can also lead to the extracellular leakage causing amplification of the inflammatory signals and production of protective autoantibodies (lower panel). See text for details. Abbreviations: cGAS, cyclic GMP-AMP synthase; IRF3, interferon regulatory factor 3; ISG, interferon stimulated genes; STING, stimulator of interferon genes; TBK1, TANK-binding kinase.
From a structural point of view, IFI16 harbors an N-terminal PYD and two C-terminal HIN200 domains (see Figure 1 for details). While AIM2 uses its PYD to interact with the inflammasome component ASC, which also contains a PYD (31, 33, 68), the direct interaction between IFI16 and ASC is still matter of debate. Nevertheless, IFI16 has been reported to induce ASC-dependent inflammasome activation during infection with some nuclear DNA viruses (32, 132, 133). Following viral DNA recognition in the nucleus, the IFI16-ASC-procaspase-1 inflammasome formation is induced. The complex is then released in the cytoplasm, where processing of pro-IL-1β into active IL-1β occurs.
Moreover, IFI16 is also an inducer of IFN-β in response to intracellular DNA. RNA interference-mediated depletion of IFI16 or its presumed mouse ortholog p204 has revealed that both proteins are required for a functional IFN response to transfected dsDNA or infection with HSV-1 in various cell types, including human and mouse monocytic cell lines (134), mouse corneal epithelial cells (135), human primary and immortalized fibroblasts (136, 137), human primary macrophages (138), neutrophils (139), and dendritic cells (140). In this regard, IFI16 has been shown to interact with STING, leading to phosphorylation and nuclear translocation of IRF3 via the IFI16–STING–TBK signaling axis, resulting in IFN-β production during HSV-1 infection (137). Moreover, IRF3 activation has been also demonstrated following direct cooperation between IFI16 and cGAS, by a mechanism in which cGAS promotes IFI16 stability in response to incoming nuclear HSV DNA, rather than through the production of 2′,3′-cGAMP (141) (Figure 3).
As aforementioned, IFI16 is unique among DNA sensors as it shuttles between the nucleus and the cytoplasm and is predominantly nuclear at steady state. Furthermore, IFI16 is able to sense DNA in both compartments depending on the localization of its DNA ligands (134, 137, 138). Thus, the ability of IFI16 to detect DNA viruses, such as HSV-1 in the nucleus, appears to contradict the long-held assumption that foreign DNA is sensed merely because of its cytosolic localization. Interestingly, the conserved HIN200 domains of the IFI16 protein are responsible for the interaction with oligonucleotide/oligosaccharide moieties (142). To what extent IFI16/p204 is involved in the sensing of DNA during infection with viruses or intracellular bacteria in vivo, and what domains are indispensable for recognition, awaits the generation of mice lacking this receptor. However, the structural studies elucidating the DNA binding of IFI16 have improved our understanding on non-self-DNA sensing and IFI16 localization. Few more issues concerning the nuclear/cytosolic interaction of STING and IFI16 and activation of inflammasome remain unanswered, mainly related to the different cellular and infection models analyzed so far.
IFN-inducible protein 16 has also been described to play a role in the DNA damage response (143, 144) and promote apoptosis and senescence (145–148). Recent reports have implicated IFI16 in autoimmunity, pointing to a role of PYHIN proteins in the pathogenesis of human autoimmune disease. Since the IFN system is largely regarded as playing a key role in autoimmune disorders including SLE, SSc, and SjS (75, 149, 150), it is possible to hypothesize that also PYHIN may play a causative role in autoimmunity thanks to its ability to induce apoptosis and trigger an inflammatory response (Figure 3). It follows that during systemic autoimmune conditions of tissue injury and apoptosis the exposure of autoantigens leads inevitably to breaking of tolerance and dysregulation of the immune response. Under physiological conditions, dead cells and tissue debris are normally cleared by the monocyte/macrophage system. However, under conditions that hamper clearance of apoptotic bodies by phagocytes, chronic exposure of autoantigens, including PYHIN proteins, may lead to the development of autoimmunity. Consistent with this scenario, various autoantigens and corresponding autoantibodies have been identified in the sera of patients affected by different systemic autoimmune diseases, such as SLE, SjS, and SSc.
Novel Functions for IFI16 to Trigger Inflammation
We have previously demonstrated that IFI16 is a key component for the tight regulation of cellular and viral promoters, through physical interaction with the nuclear transcription factor Sp1 and regulation of NF-κB pathway (151, 152). As inducer of pro-inflammatory molecules (e.g., ICAM-1, RANTES, and CCL20) and apoptosis in primary endothelial cells, IFI16 might be active during the initial phases of the inflammatory processes paving the way to the onset of autoimmunity (145). In addition, IFI16 has been shown to translocate in the cytoplasm of normal keratinocytes following UVB-induced cell injury and be subsequently released in the extracellular milieu (104). In vivo, the expression of IFI16 is significantly increased in all layers of the epidermis from patients affected by SLE or SSc, whereas in the epidermis from healthy control subjects IFI16 expression is only found in the basal layer. In the same setting, the dermal inflammatory infiltrate has been found positive for IFI16 staining indicating that IFI16 is aberrantly expressed also in lymphocytes, fibroblasts, and endothelial cells. Similarly, we and others have also recently demonstrated that IFI16 is aberrantly expressed in the intestinal mucosa of patients affected by IBD, where dysregulation of host–microbial interactions has been shown to play a major pathogenic role (153, 154). In addition, we and others have evaluated the etiopathogenic role of PYHIN proteins in the development of SLE in human pathology as well as in mouse models (Table 1). In this regard, we have found that IFI16 overexpression in primary human umbilical vein endothelial cells (HUVECs) efficiently inhibits tube morphogenesis in vitro, triggers production of pro-inflammatory molecules and leads to cell death by apoptosis, suggesting that IFI16 might induce inflammation along with other detrimental cellular pathways primarily involved in autoimmunity (145, 155).
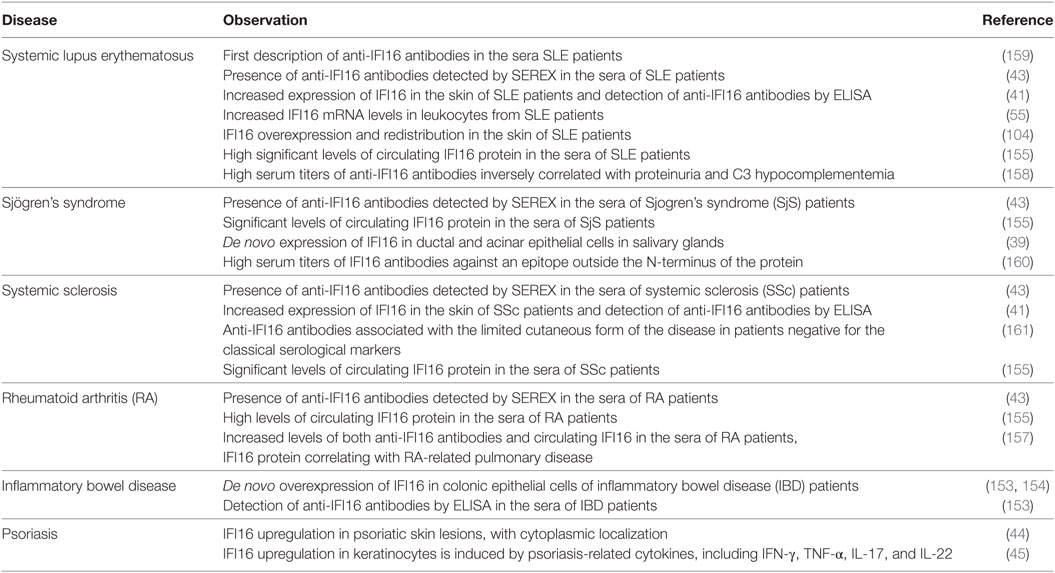
Table 1. Summary of IFN-inducible protein 16 (IFI16) correlations with systemic lupus erythematosus (SLE) and other autoimmune diseases.
In another context, IFI16 has been shown to restrict human cytomegalovirus (HCMV) and papillomavirus replication through different mechanisms (152, 156). Interestingly, IFI16 has been observed entrapped in HCMV virions undergoing cell egression (116). Consistent with our results, Singh et al. have demonstrated that IFI16 is aberrantly expressed in the cytoplasm of KSHV latently infected cells, wrapped up in exosomes and then released extracellularly (133). However, since IFI16 was originally identified as a molecule localized in intracellular compartments, in particular the nucleus, all studies on IFI16 were subsequently limited to determine the biological and physiological activity of this protein exclusively within the cellular compartment, thus disregarding a possible role of extracellular IFI16 as pro-inflammatory trigger. To fill this gap, we sought to determine the effects of extracellular IFI16 protein on HUVECs. Surprisingly, we observed a cytokine-stimulating activity of extracellular IFI16 (rIFI16) on primary endothelial cells, which led to the production and secretion of pro-inflammatory cytokines such as IL-6, IL-8, CCL2, CCL5, and CCL20. Moreover, we found that rIFI16 protein, alone or in synergy with LPS, acted by propagating “danger signals” through a MyD88-dependent TLR pathway (126).
Altogether, these results unveil a novel function of extracellular IFI16 at the endothelial interface, which might explain the ability of this protein to induce endothelial cell activation and injury during systemic inflammation.
In summary, IFI16 can promote inflammation by (1) acting as regulator of transcription factors to activate expression of genes encoding pro-inflammatory cytokines; (2) activating type I IFN production following translocation into the cytoplasm; and (3) binding to cells such as endothelial cells and keratinocytes, once released in the extracellular milieu, to activate production of pro-inflammatory chemokines and cytokines. Concomitantly, IFI16 leakage into the extracellular milieu leads to tolerance breaking and autoantibody production.
Anti-IFI16 Antibodies and Their Relation to SLE Characteristics
We and others have previously reported the presence of anti-IFI16 antibodies in sera of patients suffering from various autoimmune diseases such as SLE, SjS, AR, SSc, and IBD (39, 41, 153, 157–161) (Table 1). Among these latter, SLE stands out as the disease where IFI16 autoantibodies have been more thoroughly characterized. This aspect is of paramount importance in view of the prognostic and diagnostic relevance of other SLE autoantibodies such as ANAs and autoantibodies against Ro/SSA and La/SSB ribonucleoproteins (162). However, not all autoantibodies seem to play a causative role in autoimmunity as autoantibodies against chromatin molecules, such as HMGB1, exert a protective effect in animal models of autoimmune disease (163). Thus, new criteria for autoantibodies classification based on both their functionality and ability to trigger or dampen immunologic disturbances are clearly needed.
With regard to IFI16, it is conceivable to hypothesize that the previously described over- or aberrant expression and mislocalization of this nuclear protein, earlier in the cytoplasm and later on in the extracellular milieu, might lead to loss of tolerance and development of anti-IFI16 antibodies, as demonstrated in skin lesions from SLE patients and in keratinocytes cultured in vitro under conditions of UVB light-induced cell injury (104).
Although the occurrence of anti-IFI16 antibodies in SLE patients has long been known, their associations with clinical and serological parameters of SLE are still under debate. To address this aspect, we have recently set out to determine the prevalence of anti-IFI16 autoantibodies in a population of SLE patients from northern Italy (158). Specifically, in a cross-sectional study, we compared anti-IFI16 antibody levels of SLE patients at various stages of disease with those of patients with non-SLE primary glomerulonephritis (GN) or healthy individuals. Remarkably, we measured significantly higher anti-IFI16 titers in SLE patients compared with both disease and control groups, and, according to cutoff levels, we were able to estimate that more than 60% of the SLE patients tested positive for anti-IFI16 autoantibodies compared with just 24% of patients with primary non-SLE GN and 5% of healthy individuals. Of note, in this SLE cohort, univariate analysis showed that autoantibodies to IFI16 were inversely associated with proteinuria, whereas multivariate analysis confirmed a reduced risk of proteinuria for anti-IFI16-positive patients despite renal function. Furthermore, an inverse association between anti-IFI16 and C3 hypocomplementemia was also observed. In this regard, the association of anti-IFI16 antibodies with reduced C3 hypocomplementemia was independent of the disease activity parameters SLEDAI and anti-dsDNA. The described inverse associations between anti-IFI16 positivity, proteinuria, and C3 hypocomplementemia, together with the observation that nephritis does not occur in other systemic autoimmune diseases characterized by high titers of anti-IFI16 antibodies such as SjS and SSc, imply that ultimately these antibodies do not play a relevant role in the pathogenesis of renal inflammation in SLE, but rather most likely prevent complement consumption. Thus, based on these findings, it is likely that the occurrence of IFI16 autoantibodies might protect from the detrimental activity of the free circulating IFI16 protein, exerting beneficial functional effects rather than pathogenic ones.
Consistent with the data obtained in SLE patients, in previous studies, we found a significant prevalence of anti-IFI16 antibodies in SSc, which was more evident in the more benign limited cutaneous form of this disease (42). More recently, we have shown that enhanced titers of anti-IFI16 in IBD patients undergoing infliximab therapy correlates with a more favorable outcome of the disease (153), which can be partly explained by the protective role exerted by these antibodies against the progression of the autoimmune process.
Conclusion and Perspectives
In the last decade, we have greatly expanded our knowledge of the relationship between aberrant innate immune response and development of autoinflammatory/autoimmune diseases such as SLE. Specifically, we now know that multiple inflammasome-induced inflammatory responses correlate with the development of SLE. In this regard, the ALR family member IFI16 has been found aberrantly expressed in various target tissues of a range of autoimmune diseases, including SLE skin, SjS salivary glands, and IBD colonic epithelium. With this scenario in mind, the occurrence of anti-IFI16 antibodies is likely due to the response of the immune system to IFI16 protein release through one of the aforementioned cell death mechanisms. Alternatively, the presence of anti-IFI16 autoantibodies could be the result of IFI16 translocation from the nucleus to the cytoplasm and, eventually, being secreted into the extracellular milieu where it is recognized by the immune system. In addition, the observation that IFI16 enhances the inflammation response against microbial patterns, such as bacterial LPS, is highly suggestive of a role of ALRs also in non-canonical inflammasome-mediated signaling.
Overall, understanding the role of ALRs in SLE pathogenesis and chronic inflammation would contribute to the development of novel therapeutic options, which may not only be limited to the treatment of patients affected by systemic autoimmune disease but also to cure conditions in which prolonged inflammatory flares progressively lead to organ-specific disorders (e.g., cancer).
Author Contributions
VC, SL, MG, and MDA have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Marcello Arsura for critically reviewing the manuscript.
Funding
This work was supported by grants from the Compagnia di San Paolo (CSP2016 to MDA), the European Crohn’s and Colitis Organisation—ECCO (Research Grant 2017 to MDA), the Italian Ministry of Education, University and Research—MIUR (PRIN 2015 to MDA, 2015W729WH), the University of Turin (Research Funding 2017 to SL and MDA), and Associazione Italiana per la Ricerca sul Cancro—AIRC (IG2016 to MG).
References
1. Mitchell G, Isberg RR. Innate immunity to intracellular pathogens: balancing microbial elimination and inflammation. Cell Host Microbe (2017) 22:166–75. doi:10.1016/j.chom.2017.07.005
2. Miyake K, Kaisho T. Homeostatic inflammation in innate immunity. Curr Opin Immunol (2014) 30:85–90. doi:10.1016/j.coi.2014.08.003
3. Mantovani A. Wandering pathways in the regulation of innate immunity and inflammation. J Autoimmun (2017) 85:1–5. doi:10.1016/j.jaut.2017.10.007
4. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi:10.1016/j.cell.2010.01.022
5. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16:407–20. doi:10.1038/nri.2016.58
6. Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol (2012) 28:137–61. doi:10.1146/annurev-cellbio-101011-155745
7. Keating SE, Baran M, Bowie AG. Cytosolic DNA sensors regulating type I interferon induction. Trends Immunol (2011) 32:574–81. doi:10.1016/j.it.2011.08.004
8. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol (2013) 13:397–411. doi:10.1038/nri3452
9. Muñoz-Wolf N, Lavelle EC. Innate immune receptors. Methods Mol Biol (2016) 1417:1–43. doi:10.1007/978-1-4939-3566-6_1
10. Ratsimandresy RA, Dorfleutner A, Stehlik C. An update on PYRIN domain-containing pattern recognition receptors: from immunity to pathology. Mol Innate Immun (2013) 4:440. doi:10.3389/fimmu.2013.00440
11. Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, et al. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature (2012) 490:288–91. doi:10.1038/nature11419
12. Yi Y-S. Caspase-11 non-canonical inflammasome: a critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology (2017) 152:207–17. doi:10.1111/imm.12787
13. Hedrich CM. Shaping the spectrum – from autoinflammation to autoimmunity. Clin Immunol (2016) 165:21–8. doi:10.1016/j.clim.2016.03.002
14. Muñoz LE, Janko C, Schulze C, Schorn C, Sarter K, Schett G, et al. Autoimmunity and chronic inflammation – two clearance-related steps in the etiopathogenesis of SLE. Autoimmun Rev (2010) 10:38–42. doi:10.1016/j.autrev.2010.08.015
15. Sudres M, Verdier J, Truffault F, Le Panse R, Berrih-Aknin S. Pathophysiological mechanisms of autoimmunity. Ann N Y Acad Sci (2018) 1413:59–68. doi:10.1111/nyas.13560
16. Lisnevskaia L, Murphy G, Isenberg D. Systemic lupus erythematosus. Lancet (2014) 384:1878–88. doi:10.1016/S0140-6736(14)60128-8
17. Mohan C, Putterman C. Genetics and pathogenesis of systemic lupus erythematosus and lupus nephritis. Nat Rev Nephrol (2015) 11:329–41. doi:10.1038/nrneph.2015.33
18. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol (2010) 11:1136–42. doi:10.1038/ni.1960
19. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol (2004) 5:730–7. doi:10.1038/ni1087
20. Kolakofsky D, Kowalinski E, Cusack S. A structure-based model of RIG-I activation. RNA (2012) 18:2118–27. doi:10.1261/rna.035949.112
21. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol (2005) 6:981–8. doi:10.1038/ni1243
22. Arnoult D, Soares F, Tattoli I, Girardin SE. Mitochondria in innate immunity. EMBO Rep (2011) 12:901–10. doi:10.1038/embor.2011.157
23. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479:117–21. doi:10.1038/nature10558
24. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514:187–92. doi:10.1038/nature13683
25. Yang J, Zhao Y, Shao F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol (2015) 32:78–83. doi:10.1016/j.coi.2015.01.007
26. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science (2013) 341:1246–9. doi:10.1126/science.1240248
27. Viganò E, Diamond CE, Spreafico R, Balachander A, Sobota RM, Mortellaro A. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat Commun (2015) 6:8761. doi:10.1038/ncomms9761
28. Wacker MA, Teghanemt A, Weiss JP, Barker JH. High-affinity caspase-4 binding to LPS presented as high molecular mass aggregates or in outer membrane vesicles. Innate Immun (2017) 23:336–44. doi:10.1177/1753425917695446
29. Pellegrini C, Antonioli L, Lopez-Castejon G, Blandizzi C, Fornai M. Canonical and non-canonical activation of NLRP3 inflammasome at the crossroad between immune tolerance and intestinal inflammation. Front Immunol (2017) 8:36. doi:10.3389/fimmu.2017.00036
30. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science (2013) 341:1250–3. doi:10.1126/science.1240988
31. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458:514–8. doi:10.1038/nature07725
32. Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe (2011) 9:363–75. doi:10.1016/j.chom.2011.04.008
33. Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol (2009) 10:266–72. doi:10.1038/ni.1702
34. Albrecht M, Choubey D, Lengauer T. The HIN domain of IFI-200 proteins consists of two OB folds. Biochem Biophys Res Commun (2005) 327:679–87. doi:10.1016/j.bbrc.2004.12.056
35. Ni X, Ru H, Ma F, Zhao L, Shaw N, Feng Y, et al. New insights into the structural basis of DNA recognition by HINa and HINb domains of IFI16. J Mol Cell Biol (2016) 8:51–61. doi:10.1093/jmcb/mjv053
36. Shaw N, Liu Z-J. Role of the HIN domain in regulation of innate immune responses. Mol Cell Biol (2014) 34:2–15. doi:10.1128/MCB.00857-13
37. Xiao TS. The nucleic acid-sensing inflammasomes. Immunol Rev (2015) 265:103–11. doi:10.1111/imr.12281
38. Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity (2012) 36:561–71. doi:10.1016/j.immuni.2012.02.014
39. Alunno A, Caneparo V, Carubbi F, Bistoni O, Caterbi S, Bartoloni E, et al. Interferon gamma-inducible protein 16 in primary Sjögren’s syndrome: a novel player in disease pathogenesis? Arthritis Res Ther (2015) 17:208. doi:10.1186/s13075-015-0722-2
40. Choubey D. Interferon-inducible Ifi200-family genes as modifiers of lupus susceptibility. Immunol Lett (2012) 147:10–7. doi:10.1016/j.imlet.2012.07.003
41. Mondini M, Vidali M, De Andrea M, Azzimonti B, Airò P, D’Ambrosio R, et al. A novel autoantigen to differentiate limited cutaneous systemic sclerosis from diffuse cutaneous systemic sclerosis: the interferon-inducible gene IFI16. Arthritis Rheum (2006) 54:3939–44. doi:10.1002/art.22266
42. Mondini M, Vidali M, Airò P, De Andrea M, Riboldi P, Meroni PL, et al. Role of the interferon-inducible gene IFI16 in the etiopathogenesis of systemic autoimmune disorders. Ann N Y Acad Sci (2007) 1110:47–56. doi:10.1196/annals.1423.006
43. Uchida K, Akita Y, Matsuo K, Fujiwara S, Nakagawa A, Kazaoka Y, et al. Identification of specific autoantigens in Sjögren’s syndrome by SEREX. Immunology (2005) 116:53–63. doi:10.1111/j.1365-2567.2005.02197.x
44. Chiliveru S, Rahbek SH, Jensen SK, Jørgensen SE, Nissen SK, Christiansen SH, et al. Inflammatory cytokines break down intrinsic immunological tolerance of human primary keratinocytes to cytosolic DNA. J Immunol (2014) 192:2395–404. doi:10.4049/jimmunol.1302120
45. Cao T, Shao S, Li B, Jin L, Lei J, Qiao H, et al. Up-regulation of Interferon-inducible protein 16 contributes to psoriasis by modulating chemokine production in keratinocytes. Sci Rep (2016) 6:25381. doi:10.1038/srep25381
46. Borchers AT, Leibushor N, Naguwa SM, Cheema GS, Shoenfeld Y, Gershwin ME. Lupus nephritis: a critical review. Autoimmun Rev (2012) 12:174–94. doi:10.1016/j.autrev.2012.08.018
47. Herszényi L, Tulassay Z. The role of autoantibodies in inflammatory bowel disease. Dig Dis (2012) 30:201–7. doi:10.1159/000336702
48. Yang C-A, Chiang B-L. Inflammasomes and human autoimmunity: a comprehensive review. J Autoimmun (2015) 61:1–8. doi:10.1016/j.jaut.2015.05.001
49. Choubey D, Panchanathan R. Absent in melanoma 2 proteins in SLE. Clin Immunol (2017) 176:42–8. doi:10.1016/j.clim.2016.12.011
50. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature (2010) 464:1357–61. doi:10.1038/nature08938
51. Inoue M, Shinohara ML. NLRP3 Inflammasome and MS/EAE. Autoimmune Dis (2013) 2013:859145. doi:10.1155/2013/859145
52. Kastbom A, Verma D, Eriksson P, Skogh T, Wingren G, Söderkvist P. Genetic variation in proteins of the cryopyrin inflammasome influences susceptibility and severity of rheumatoid arthritis (the Swedish TIRA project). Rheumatology (Oxford) (2008) 47:415–7. doi:10.1093/rheumatology/kem372
53. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti T-D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity (2010) 32:379–91. doi:10.1016/j.immuni.2010.03.003
54. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol (2010) 11:136–40. doi:10.1038/ni.1831
55. Kimkong I, Avihingsanon Y, Hirankarn N. Expression profile of HIN200 in leukocytes and renal biopsy of SLE patients by real-time RT-PCR. Lupus (2009) 18:1066–72. doi:10.1177/0961203309106699
56. Cagliani R, Forni D, Biasin M, Comabella M, Guerini FR, Riva S, et al. Ancient and recent selective pressures shaped genetic diversity at AIM2-like nucleic acid sensors. Genome Biol Evol (2014) 6:830–45. doi:10.1093/gbe/evu066
57. Jin Y, Mailloux CM, Gowan K, Riccardi SL, LaBerge G, Bennett DC, et al. NALP1 in vitiligo-associated multiple autoimmune disease. N Engl J Med (2007) 356:1216–25. doi:10.1056/NEJMoa061592
58. Ortiz-Fernández L, García-Lozano J-R, Montes-Cano M-A, Conde-Jaldón M, Ortego-Centeno N, García-Hernández F-J, et al. Variants of the IFI16 gene affecting the levels of expression of mRNA are associated with susceptibility to Behçet disease. J Rheumatol (2015) 42:695–701. doi:10.3899/jrheum.140949
59. Pontillo A, Girardelli M, Kamada AJ, Pancotto JAT, Donadi EA, Crovella S, et al. Polymorphisms in inflammasome genes are involved in the predisposition to systemic lupus erythematosus. Autoimmunity (2012) 45:271–8. doi:10.3109/08916934.2011.637532
60. Verma D, Särndahl E, Andersson H, Eriksson P, Fredrikson M, Jönsson J-I, et al. The Q705K polymorphism in NLRP3 is a gain-of-function alteration leading to excessive interleukin-1β and IL-18 production. PLoS One (2012) 7:e34977. doi:10.1371/journal.pone.0034977
61. Yang C-A, Huang S-T, Chiang B-L. Sex-dependent differential activation of NLRP3 and AIM2 inflammasomes in SLE macrophages. Rheumatology (Oxford) (2015) 54:324–31. doi:10.1093/rheumatology/keu318
62. Harrison P, Pointon JJ, Chapman K, Roddam A, Wordsworth BP. Interleukin-1 promoter region polymorphism role in rheumatoid arthritis: a meta-analysis of IL-1B-511A/G variant reveals association with rheumatoid arthritis. Rheumatology (Oxford) (2008) 47:1768–70. doi:10.1093/rheumatology/ken374
63. Karakas Celik S, Öz ZS, Dursun A, Unal A, Emre U, Cicek S, et al. Interleukin 18 gene polymorphism is a risk factor for multiple sclerosis. Mol Biol Rep (2014) 41:1653–8. doi:10.1007/s11033-013-3013-5
64. Wen D, Liu J, Du X, Dong J-Z, Ma C-S. Association of interleukin-18 (-137G/C) polymorphism with rheumatoid arthritis and systemic lupus erythematosus: a meta-analysis. Int Rev Immunol (2014) 33:34–44. doi:10.3109/08830185.2013.816699
65. Wang CQF, Cruz-Inigo AE, Fuentes-Duculan J, Moussai D, Gulati N, Sullivan-Whalen M, et al. Th17 cells and activated dendritic cells are increased in vitiligo lesions. PLoS One (2011) 6:e18907. doi:10.1371/journal.pone.0018907
66. Choulaki C, Papadaki G, Repa A, Kampouraki E, Kambas K, Ritis K, et al. Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res Ther (2015) 17:257. doi:10.1186/s13075-015-0775-2
67. Sørensen TL, Sellebjerg F. Distinct chemokine receptor and cytokine expression profile in secondary progressive MS. Neurology (2001) 57:1371–6. doi:10.1212/WNL.57.8.1371
68. Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458:509–13. doi:10.1038/nature07710
69. Zhang W, Cai Y, Xu W, Yin Z, Gao X, Xiong S. AIM2 facilitates the apoptotic DNA-induced systemic lupus erythematosus via arbitrating macrophage functional maturation. J Clin Immunol (2013) 33:925–37. doi:10.1007/s10875-013-9881-6
70. Eloranta M-L, Alm GV, Rönnblom L. Disease mechanisms in rheumatology – tools and pathways: plasmacytoid dendritic cells and their role in autoimmune rheumatic diseases. Arthritis Rheum (2013) 65:853–63. doi:10.1002/art.37821
71. Olsen NJ, Karp DR. Autoantibodies and SLE: the threshold for disease. Nat Rev Rheumatol (2014) 10:181–6. doi:10.1038/nrrheum.2013.184
72. Bai Y, Tong Y, Liu Y, Hu H. Self-dsDNA in the pathogenesis of systemic lupus erythematosus. Clin Exp Immunol (2018) 191:1–10. doi:10.1111/cei.13041
73. Rekvig OP. Anti-dsDNA antibodies as a classification criterion and a diagnostic marker for systemic lupus erythematosus: critical remarks. Clin Exp Immunol (2015) 179:5–10. doi:10.1111/cei.12296
74. Rönnblom L, Eloranta M-L. The interferon signature in autoimmune diseases. Curr Opin Rheumatol (2013) 25:248–53. doi:10.1097/BOR.0b013e32835c7e32
75. Crow MK. Advances in understanding the role of type I interferons in systemic lupus erythematosus. Curr Opin Rheumatol (2014) 26:467–74. doi:10.1097/BOR.0000000000000087
76. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med (2011) 3:73ra19. doi:10.1126/scitranslmed.3001180
77. Muñoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol (2010) 6:280–9. doi:10.1038/nrrheum.2010.46
78. Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum (2002) 46:191–201. doi:10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K
79. Boswell JM, Yui MA, Burt DW, Kelley VE. Increased tumor necrosis factor and IL-1 beta gene expression in the kidneys of mice with lupus nephritis. J Immunol (1988) 141:3050–4.
80. Wang B, Zhu J-M, Fan Y-G, Feng C-C, Chen G-M, Chen H, et al. The association of IL1α and IL1β polymorphisms with susceptibility to systemic lupus erythematosus: a meta-analysis. Gene (2013) 527:95–101. doi:10.1016/j.gene.2013.05.059
81. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ (2018) 25(3):486–541. doi:10.1038/s41418-017-0012-4
82. Mahajan A, Herrmann M, Muñoz LE. Clearance deficiency and cell death pathways: a model for the pathogenesis of SLE. Front Immunol (2016) 7:35. doi:10.3389/fimmu.2016.00035
83. Magna M, Pisetsky DS. The role of cell death in the pathogenesis of sle: is pyroptosis the missing link? Scand J Immunol (2015) 82:218–24. doi:10.1111/sji.12335
84. Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol (2008) 9:231–41. doi:10.1038/nrm2312
85. Biermann M, Maueröder C, Brauner JM, Chaurio R, Janko C, Herrmann M, et al. Surface code – biophysical signals for apoptotic cell clearance. Phys Biol (2013) 10:065007. doi:10.1088/1478-3975/10/6/065007
86. Silva MT. Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett (2010) 584:4491–9. doi:10.1016/j.febslet.2010.10.046
87. Anam K, Amare M, Naik S, Szabo KA, Davis TA. Severe tissue trauma triggers the autoimmune state systemic lupus erythematosus in the MRL/++ lupus-prone mouse. Lupus (2009) 18:318–31. doi:10.1177/0961203308097479
88. Rigante D, Mazzoni MB, Esposito S. The cryptic interplay between systemic lupus erythematosus and infections. Autoimmun Rev (2014) 13:96–102. doi:10.1016/j.autrev.2013.09.004
89. Kazzaz NM, Sule G, Knight JS. Intercellular interactions as regulators of NETosis. Front Immunol (2016) 7:453. doi:10.3389/fimmu.2016.00453
90. Boe DM, Curtis BJ, Chen MM, Ippolito JA, Kovacs EJ. Extracellular traps and macrophages: new roles for the versatile phagocyte. J Leukoc Biol (2015) 97:1023–35. doi:10.1189/jlb.4RI1014-521R
91. Farrera C, Fadeel B. Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol (2013) 191:2647–56. doi:10.4049/jimmunol.1300436
92. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol (2011) 187:538–52. doi:10.4049/jimmunol.1100450
93. Kienhöfer D, Hahn J, Stoof J, Csepregi JZ, Reinwald C, Urbonaviciute V, et al. Experimental lupus is aggravated in mouse strains with impaired induction of neutrophil extracellular traps. JCI Insight (2017) 2:92920. doi:10.1172/jci.insight.92920
94. Boyapati RK, Tamborska A, Dorward DA, Ho G-T. Advances in the understanding of mitochondrial DNA as a pathogenic factor in inflammatory diseases. F1000Res (2017) 6:169. doi:10.12688/f1000research.10397.1
95. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med (2016) 22:146–53. doi:10.1038/nm.4027
96. Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med (2016) 213:697–713. doi:10.1084/jem.20151876
97. Wang H, Li T, Chen S, Gu Y, Ye S. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol (2015) 67:3190–200. doi:10.1002/art.39296
98. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature (2009) 461:788–92. doi:10.1038/nature08476
99. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2013) 339:786–91. doi:10.1126/science.1232458
100. Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J Biol Chem (2007) 282:15325–9. doi:10.1074/jbc.R700002200
101. Lehmann P, Homey B. Clinic and pathophysiology of photosensitivity in lupus erythematosus. Autoimmun Rev (2009) 8:456–61. doi:10.1016/j.autrev.2008.12.012
102. Wenzel J, Tüting T. Identification of type I interferon-associated inflammation in the pathogenesis of cutaneous lupus erythematosus opens up options for novel therapeutic approaches. Exp Dermatol (2007) 16:454–63. doi:10.1111/j.1600-0625.2007.00556.x
103. Reefman E, Kuiper H, Limburg PC, Kallenberg CGM, Bijl M. Type I interferons are involved in the development of ultraviolet B-induced inflammatory skin lesions in systemic lupus erythaematosus patients. Ann Rheum Dis (2008) 67:11–8. doi:10.1136/ard.2007.070359
104. Costa S, Borgogna C, Mondini M, De Andrea M, Meroni PL, Berti E, et al. Redistribution of the nuclear protein IFI16 into the cytoplasm of ultraviolet B-exposed keratinocytes as a mechanism of autoantigen processing. Br J Dermatol (2011) 164:282–90. doi:10.1111/j.1365-2133.2010.10097.x
105. Kemp MG, Lindsey-Boltz LA, Sancar A. UV light potentiates STING (stimulator of interferon genes)-dependent innate immune signaling through deregulation of ULK1 (Unc51-like kinase 1). J Biol Chem (2015) 290:12184–94. doi:10.1074/jbc.M115.649301
106. Mu Q, Zhang H, Luo XM. SLE: another autoimmune disorder influenced by microbes and diet? Front Immunol (2015) 6:608. doi:10.3389/fimmu.2015.00608
107. Nelson P, Rylance P, Roden D, Trela M, Tugnet N. Viruses as potential pathogenic agents in systemic lupus erythematosus. Lupus (2014) 23:596–605. doi:10.1177/0961203314531637
108. Brencicova E, Diebold SS. Nucleic acids and endosomal pattern recognition: how to tell friend from foe? Front Cell Infect Microbiol (2013) 3:37. doi:10.3389/fcimb.2013.00037
109. Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol (2014) 32:461–88. doi:10.1146/annurev-immunol-032713-120156
110. Chan YK, Gack MU. RIG-I-like receptor regulation in virus infection and immunity. Curr Opin Virol (2015) 12:7–14. doi:10.1016/j.coviro.2015.01.004
111. Reikine S, Nguyen JB, Modis Y. Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front Immunol (2014) 5:342. doi:10.3389/fimmu.2014.00342
112. Liu Y, Olagnier D, Lin R. Host and viral modulation of RIG-I-mediated antiviral immunity. Front Immunol (2016) 7:662. doi:10.3389/fimmu.2016.00662
113. Luecke S, Paludan SR. Molecular requirements for sensing of intracellular microbial nucleic acids by the innate immune system. Cytokine (2017) 98:4–14. doi:10.1016/j.cyto.2016.10.003
114. Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature (2007) 448:501–5. doi:10.1038/nature06013
115. Lugrin J, Martinon F. The AIM2 inflammasome: sensor of pathogens and cellular perturbations. Immunol Rev (2018) 281:99–114. doi:10.1111/imr.12618
116. Dell’Oste V, Gatti D, Gugliesi F, De Andrea M, Bawadekar M, Lo Cigno I, et al. Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage of in vitro human cytomegalovirus infection and is entrapped in the egressing virions during the late stage. J Virol (2014) 88:6970–82. doi:10.1128/JVI.00384-14
117. Jakobsen MR, Paludan SR. IFI16: at the interphase between innate DNA sensing and genome regulation. Cytokine Growth Factor Rev (2014) 25:649–55. doi:10.1016/j.cytogfr.2014.06.004
118. Barbé F, Douglas T, Saleh M. Advances in Nod-like receptors (NLR) biology. Cytokine Growth Factor Rev (2014) 25:681–97. doi:10.1016/j.cytogfr.2014.07.001
119. Jiang Y, Zhu Y, Liu Z-J, Ouyang S. The emerging roles of the DDX41 protein in immunity and diseases. Protein Cell (2017) 8:83–9. doi:10.1007/s13238-016-0303-4
120. Ori D, Murase M, Kawai T. Cytosolic nucleic acid sensors and innate immune regulation. Int Rev Immunol (2017) 36:74–88. doi:10.1080/08830185.2017.1298749
121. Barrat FJ, Elkon KB, Fitzgerald KA. Importance of nucleic acid recognition in inflammation and autoimmunity. Annu Rev Med (2016) 67:323–36. doi:10.1146/annurev-med-052814-023338
122. Crowl JT, Gray EE, Pestal K, Volkman HE, Stetson DB. Intracellular nucleic acid detection in autoimmunity. Annu Rev Immunol (2017) 35:313–36. doi:10.1146/annurev-immunol-051116-052331
123. Gay NJ, Gangloff M, O’Neill LAJ. What the myddosome structure tells us about the initiation of innate immunity. Trends Immunol (2011) 32:104–9. doi:10.1016/j.it.2010.12.005
124. Malik A, Kanneganti T-D. Inflammasome activation and assembly at a glance. J Cell Sci (2017) 130:3955–63. doi:10.1242/jcs.207365
125. Asefa B, Klarmann KD, Copeland NG, Gilbert DJ, Jenkins NA, Keller JR. The interferon-inducible p200 family of proteins: a perspective on their roles in cell cycle regulation and differentiation. Blood Cells Mol Dis (2004) 32:155–67. doi:10.1016/j.bcmd.2003.10.002
126. Bawadekar M, De Andrea M, Lo Cigno I, Baldanzi G, Caneparo V, Graziani A, et al. The extracellular IFI16 protein propagates inflammation in endothelial cells via p38 MAPK and NF-κB p65 activation. J Interferon Cytokine Res (2015) 35:441–53. doi:10.1089/jir.2014.0168
127. Choubey D. Absent in melanoma 2 proteins in the development of cancer. Cell Mol Life Sci (2016) 73:4383–95. doi:10.1007/s00018-016-2296-9
128. Gariglio M, Mondini M, De Andrea M, Landolfo S. The multifaceted interferon-inducible p200 family proteins: from cell biology to human pathology. J Interferon Cytokine Res (2011) 31:159–72. doi:10.1089/jir.2010.0106
129. Man SM, Karki R, Kanneganti T-D. AIM2 inflammasome in infection, cancer, and autoimmunity: role in DNA sensing, inflammation, and innate immunity. Eur J Immunol (2016) 46:269–80. doi:10.1002/eji.201545839
130. Xin H, Curry J, Johnstone RW, Nickoloff BJ, Choubey D. Role of IFI 16, a member of the interferon-inducible p200-protein family, in prostate epithelial cellular senescence. Oncogene (2003) 22:4831–40. doi:10.1038/sj.onc.1206754
131. Gariglio M, Azzimonti B, Pagano M, Palestro G, De Andrea M, Valente G, et al. Immunohistochemical expression analysis of the human interferon-inducible gene IFI16, a member of the HIN200 family, not restricted to hematopoietic cells. J Interferon Cytokine Res (2002) 22:815–21. doi:10.1089/107999002320271413
132. Ansari MA, Singh VV, Dutta S, Veettil MV, Dutta D, Chikoti L, et al. Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells. J Virol (2013) 87:8606–23. doi:10.1128/JVI.00805-13
133. Singh VV, Kerur N, Bottero V, Dutta S, Chakraborty S, Ansari MA, et al. Kaposi’s sarcoma-associated herpesvirus latency in endothelial and B cells activates gamma interferon-inducible protein 16-mediated inflammasomes. J Virol (2013) 87:4417–31. doi:10.1128/JVI.03282-12
134. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol (2010) 11:997–1004. doi:10.1038/ni.1932
135. Conrady CD, Zheng M, Fitzgerald KA, Liu C, Carr DJJ. Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal Immunol (2012) 5:173–83. doi:10.1038/mi.2011.63
136. Duan X, Ponomareva L, Veeranki S, Choubey D. IFI16 induction by glucose restriction in human fibroblasts contributes to autophagy through activation of the ATM/AMPK/p53 pathway. PLoS One (2011) 6:e19532. doi:10.1371/journal.pone.0019532
137. Orzalli MH, DeLuca NA, Knipe DM. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A (2012) 109:E3008–17. doi:10.1073/pnas.1211302109
138. Horan KA, Hansen K, Jakobsen MR, Holm CK, Søby S, Unterholzner L, et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J Immunol (2013) 190:2311–9. doi:10.4049/jimmunol.1202749
139. Tamassia N, Bazzoni F, Le Moigne V, Calzetti F, Masala C, Grisendi G, et al. IFN-β expression is directly activated in human neutrophils transfected with plasmid DNA and is further increased via TLR-4-mediated signaling. J Immunol (2012) 189:1500–9. doi:10.4049/jimmunol.1102985
140. Kis-Toth K, Szanto A, Thai T-H, Tsokos GC. Cytosolic DNA-activated human dendritic cells are potent activators of the adaptive immune response. J Immunol (2011) 187:1222–34. doi:10.4049/jimmunol.1100469
141. Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, et al. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci U S A (2015) 112:E1773–81. doi:10.1073/pnas.1424637112
142. Hároníková L, Coufal J, Kejnovská I, Jagelská EB, Fojta M, Dvořáková P, et al. IFI16 preferentially binds to DNA with quadruplex structure and enhances DNA quadruplex formation. PLoS One (2016) 11:e0157156. doi:10.1371/journal.pone.0157156
143. Fujiuchi N, Aglipay JA, Ohtsuka T, Maehara N, Sahin F, Su GH, et al. Requirement of IFI16 for the maximal activation of p53 induced by ionizing radiation. J Biol Chem (2004) 279:20339–44. doi:10.1074/jbc.M400344200
144. Liao JCC, Lam R, Brazda V, Duan S, Ravichandran M, Ma J, et al. Interferon-inducible protein 16: insight into the interaction with tumor suppressor p53. Structure (2011) 19:418–29. doi:10.1016/j.str.2010.12.015
145. Gugliesi F, De Andrea M, Mondini M, Cappello P, Giovarelli M, Shoenfeld Y, et al. The proapoptotic activity of the interferon-inducible gene IFI16 provides new insights into its etiopathogenetic role in autoimmunity. J Autoimmun (2010) 35:114–23. doi:10.1016/j.jaut.2010.04.001
146. Kim E-J, Park J-I, Nelkin BD. IFI16 is an essential mediator of growth inhibition, but not differentiation, induced by the leukemia inhibitory factor/JAK/STAT pathway in medullary thyroid carcinoma cells. J Biol Chem (2005) 280:4913–20. doi:10.1074/jbc.M410542200
147. Song LL, Ponomareva L, Shen H, Duan X, Alimirah F, Choubey D. Interferon-inducible IFI16, a negative regulator of cell growth, down-regulates expression of human telomerase reverse transcriptase (hTERT) gene. PLoS One (2010) 5:e8569. doi:10.1371/journal.pone.0008569
148. Zhang Y, Howell RD, Alfonso DT, Yu J, Kong L, Wittig JC, et al. IFI16 inhibits tumorigenicity and cell proliferation of bone and cartilage tumor cells. Front Biosci (2007) 12:4855–63. doi:10.2741/2433
149. Wu M, Assassi S. The role of type 1 interferon in systemic sclerosis. Front Immunol (2013) 4:266. doi:10.3389/fimmu.2013.00266
150. Yao Y, Liu Z, Jallal B, Shen N, Rönnblom L. Type I interferons in Sjögren’s syndrome. Autoimmun Rev (2013) 12:558–66. doi:10.1016/j.autrev.2012.10.006
151. Caposio P, Gugliesi F, Zannetti C, Sponza S, Mondini M, Medico E, et al. A novel role of the interferon-inducible protein IFI16 as inducer of proinflammatory molecules in endothelial cells. J Biol Chem (2007) 282:33515–29. doi:10.1074/jbc.M701846200
152. Gariano GR, Dell’Oste V, Bronzini M, Gatti D, Luganini A, De Andrea M, et al. The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog (2012) 8:e1002498. doi:10.1371/journal.ppat.1002498
153. Caneparo V, Pastorelli L, Pisani LF, Bruni B, Prodam F, Boldorini R, et al. Distinct anti-IFI16 and anti-GP2 antibodies in inflammatory bowel disease and their variation with infliximab therapy. Inflamm Bowel Dis (2016) 22:2977–87. doi:10.1097/MIB.0000000000000926
154. Vanhove W, Peeters PM, Staelens D, Schraenen A, Van der Goten J, Cleynen I, et al. Strong upregulation of AIM2 and IFI16 inflammasomes in the mucosa of patients with active inflammatory bowel disease. Inflamm Bowel Dis (2015) 21:2673–82. doi:10.1097/MIB.0000000000000535
155. Gugliesi F, Bawadekar M, De Andrea M, Dell’Oste V, Caneparo V, Tincani A, et al. Nuclear DNA sensor IFI16 as circulating protein in autoimmune diseases is a signal of damage that impairs endothelial cells through high-affinity membrane binding. PLoS One (2013) 8:e63045. doi:10.1371/journal.pone.0063045
156. Lo Cigno I, De Andrea M, Borgogna C, Albertini S, Landini MM, Peretti A, et al. The nuclear DNA sensor IFI16 acts as a restriction factor for human papillomavirus replication through epigenetic modifications of the viral promoters. J Virol (2015) 89:7506–20. doi:10.1128/JVI.00013-15
157. Alunno A, Caneparo V, Bistoni O, Caterbi S, Terenzi R, Gariglio M, et al. Circulating interferon-inducible protein IFI16 correlates with clinical and serological features in rheumatoid arthritis. Arthritis Care Res (2016) 68:440–5. doi:10.1002/acr.22695
158. Caneparo V, Cena T, De Andrea M, Dell’oste V, Stratta P, Quaglia M, et al. Anti-IFI16 antibodies and their relation to disease characteristics in systemic lupus erythematosus. Lupus (2013) 22:607–13. doi:10.1177/0961203313484978
159. Seelig HP, Ehrfeld H, Renz M. Interferon-gamma-inducible protein p16. a new target of antinuclear antibodies in patients with systemic lupus erythematosus. Arthritis Rheum (1994) 37:1672–83. doi:10.1002/art.1780371117
160. Baer AN, Petri M, Sohn J, Rosen A, Casciola-Rosen L. Association of antibodies to interferon-inducible protein-16 with markers of more severe disease in primary Sjögren’s syndrome. Arthritis Care Res (2016) 68:254–60. doi:10.1002/acr.22632
161. Costa S, Mondini M, Caneparo V, Afeltra A, Airò P, Bellisai F, et al. Detection of anti-IFI16 antibodies by ELISA: clinical and serological associations in systemic sclerosis. Rheumatology (Oxford) (2011) 50:674–81. doi:10.1093/rheumatology/keq372
162. Pisetsky DS. Antinuclear antibodies in rheumatic disease: a proposal for a function-based classification. Scand J Immunol (2012) 76:223–8. doi:10.1111/j.1365-3083.2012.02728.x
Keywords: IFN-inducible protein 16, absent in melanoma 2 (AIM2)-like receptor, inflammasome, interferon, systemic lupus erythematosus
Citation: Caneparo V, Landolfo S, Gariglio M and De Andrea M (2018) The Absent in Melanoma 2-Like Receptor IFN-Inducible Protein 16 as an Inflammasome Regulator in Systemic Lupus Erythematosus: The Dark Side of Sensing Microbes. Front. Immunol. 9:1180. doi: 10.3389/fimmu.2018.01180
Received: 28 February 2018; Accepted: 11 May 2018;
Published: 28 May 2018
Edited by:
Pier Luigi Meroni, Istituto Auxologico Italiano (IRCCS), ItalyReviewed by:
James Harris, Monash University, AustraliaCaroline Jefferies, Cedars-Sinai Medical Center, United States
Copyright: © 2018 Caneparo, Landolfo, Gariglio and De Andrea. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco De Andrea, marco.deandrea@unito.it