





Abstract
The pathological hallmarks of chronic obstructive pulmonary disease (COPD) are inflammation of the small airways (bronchiolitis) and destruction of lung parenchyma (emphysema). The functional consequence of these abnormalities is airflow limitation.
Airway abnormalities and emphysema interact in a complex fashion in the development of airflow limitation in COPD. In an attempt to improve understanding of the role of the airways in COPD, the morphological counterparts of airflow limitation, the cellular inflammatory infiltrate in the airways and the relationship between emphysema type and airway abnormalities are reviewed.
Significant correlation between airway remodelling and functional measurements are found in earlier stages of COPD. In advanced COPD, airflow limitation is reflected by airway narrowing, airway deformity and extent of emphysema. The cellular inflammatory infiltrate is mainly composed of neutrophils in early stages of COPD. However, the presence of CD8+ T‐cells seems to differentiate smokers with COPD from smokers that would not develop the disease. The inflammatory changes and remodelling found in the airways and their contribution to airflow obstruction might be modulated by the type of emphysema smokers develop, with centrilobular emphysema showing more severe inflammatory changes and narrower airways than panlobular emphysema.
In conclusion, the degree of airway involvement in chronic obstructive pulmonary disease can vary greatly for the same degree of airflow obstruction, depending on the type of emphysema smokers develop. If the underlying lung abnormalities in chronic obstructive pulmonary disease vary, as the evidence suggests, the study of cigarette-induced lung disease as a single entity will further delay understanding of chronic obstructive pulmonary disease.
The pathological hallmarks of chronic obstructive pulmonary disease (COPD) are inflammation of the small airways (bronchiolitis) and destruction of lung parenchyma (emphysema). The functional consequence of these abnormalities is airflow limitation.
Bronchiolitis contributes to airflow limitation by narrowing and obliterating the airway lumen and by actively constricting the airway. Emphysema, by reducing the elastic recoil of the lung through parenchymal destruction and increase in alveolar size, as well as reducing the elastic load applied to the airways through destruction of alveolar attachments, also contributes to the airflow limitation characteristic of smokers. Although there is increasing evidence that the large airways are inflamed in patients with COPD, this is not believed to contribute directly to the airflow limitation in these patients.
It has become apparent that airway abnormalities and emphysema interact in a complex fashion, rather than simply additively in the development of airflow limitation and COPD. The type of emphysema that smokers develop seems to determine, to a great extent, the quantity and possibly type of inflammation and airway remodelling found, suggesting that different pathogenetic mechanisms might be at play in the development of COPD.
Understanding of the role of the airways in COPD might be helped by considering the different aspects of airway abnormalities covered in the present review: 1) the morphological counterparts of airflow limitation; 2) the cellular inflammatory infiltrate in the airways; and 3) the relationship between the emphysema type and airway abnormalities.
The morphological counterparts of airflow limitation
Contrary to previous opinion, Macklem and Mead 1, using the novel retrograde catheter technique demonstrated that airways of <2 mm in diameter (small airways) contributed no more than a quarter of the total airway resistance found in dog lungs. Hogg et al. 2, applying the same technique to human lungs, found that only 25% of the total airway resistance was contributed by airways of <2–3 mm in diameter in excised normal lungs. However, in smokers with mild emphysema, there was a four-fold increase in peripheral airway resistance with total airway resistance remaining unchanged. More severe degrees of emphysema resulted in a marked increase in total airway resistance due almost entirely to the increase in the peripheral airway component. This work established the then new, and still prevailing, concept that peripheral airways are the major site of increased resistance and disease in smoke-induced obstructive lung disease, and that significant increases in the resistance of these airways can be present without changes in the total airway resistance of the lung.
Niewoehner et al. 3 were the first to demonstrate that definite pathological abnormalities could already be present in the peripheral airways of young smokers. Membranous bronchioles showed a denuded epithelium and increased number of mural inflammatory cells. The most prominent finding was what they termed respiratory bronchiolitis. This lesion was characterized by clusters of pigmented macrophages in the bronchiolar lumina, frequently associated with oedema, fibrosis and epithelial hyperplasia in adjacent bronchiolar and alveolar walls. Such abnormalities were not found in the airways of nonsmokers of the same age. This study demonstrated a definite association between cigarette smoking and pathological changes in the peripheral airways, and it was hypothesized that these lesions may be responsible for the subtle physiological abnormalities observed in young smokers and may be the precursors of more severe anatomical lesions.
Morphology and function correlation
By studying smokers who had undergone tests of pulmonary function, including those reflecting the small airways, before undergoing resection for lung tumours, investigators at McGill University, Montreal, Canada, developed a pathological score to describe the microscopic changes in the small airways of smokers to study the correlations between morphology and function 4. Specifically, they scored luminal occlusion, goblet cell metaplasia, squamous cell metaplasia, mucosal ulcers, muscle hypertrophy, inflammatory cell infiltrate, fibrosis and pigment deposition of the airway wall in airways <2 mm in diameter. This study showed that these patients had similar but much more extensive abnormalities of the small airways than those described by Niewoehner et al. 3. These differences were most probably due to the fact that the patients studied at McGill University were older, had smoked more and already had some degree of COPD. The first abnormalities that could be seen in the older smokers were changes in the epithelium, with squamous and goblet cell metaplasia and a chronic inflammatory infiltrate, and a slight increase in the amount of connective tissue in the walls of the small airways. As the pathological and functional abnormalities progressed, the cellular inflammatory infiltrate changed little, but there was a progressive increase in the amount of connective tissue pigment and muscle in the airway wall (fig. 1⇓).
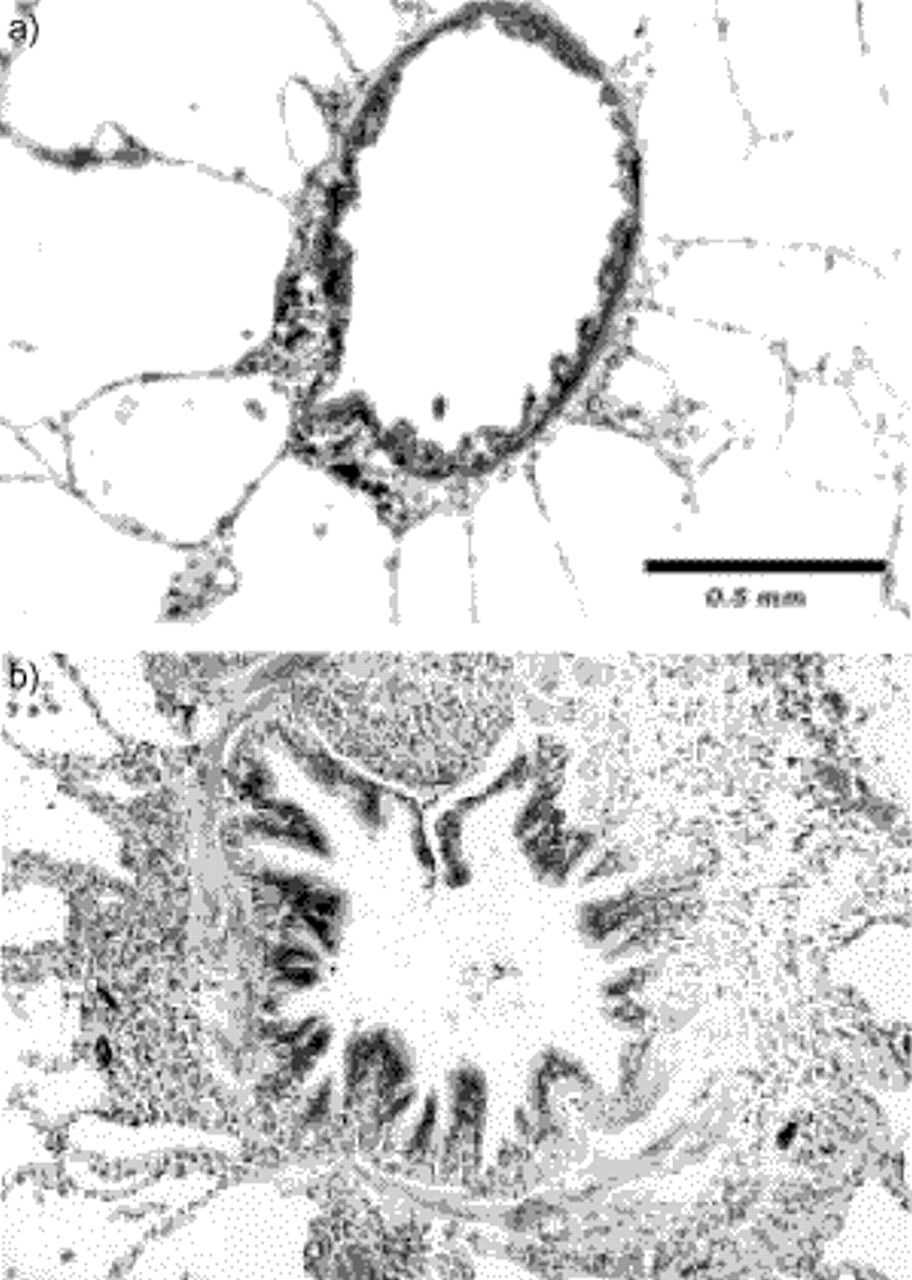
a) Normal membranous bronchiole characterized by ciliated epithelium with no goblet cells, a thin wall with a full layer of smooth muscle and numerous attached alveoli. b) Membranous bronchiole in a smoker with airflow limitation. The lumen is narrowed and deformed, the wall is thick with abundant fibrosis and muscle, and inflammatory cells and pigment deposition are apparent. Internal scale bar=0.5 mm.
When physiological measurements reflecting small airway abnormalities such as the nitrogen-washout test, volume of isoflow, change in maximal expiratory flow at the 50% vital capacity between air and helium (ΔVmax50), and other functional parameters such as the forced expiratory volume in one second (FEV1)/forced vital capacity (FVC) ratio, mid-expiratory flow rate and residual volume were compared with the pathological score, all measurements showed a progressive deterioration as the score for the morphological abnormalities increased, but only the group with the most severe small airway score demonstrated a substantial amount of emphysema. The striking correlation between the progression of physiological impairment and the degree of small airway disease suggested that inflammatory changes of the small airways made an important contribution to the functional deterioration seen in COPD even in the presence of emphysema. Furthermore, in subjects with a normal FEV1/FVC ratio, two tests of small airway function, the slope of phase III of the nitrogen washout and the volume of isoflow of the air/helium flow/volume loops, were able to detect mild abnormalities of the small airways when the results of other spirometric tests were normal 4.
Other investigators later confirmed these findings using lungs obtained either during surgery or at autopsy. Berend et al. 5 showed that the results of tests of small airway function correlated best with the total pathological score of the small airways and, in particular, with the inflammatory score. They also found that the closing volume and mid-expiratory flow correlated significantly with a measure of airway luminal size 6. Performing maximal expiratory flow volume curve experiments in excised human lungs, Berend et al. 7 also showed that flow at the trachea correlated significantly with the total pathological score of the small airways as well as the inflammation, fibrosis and emphysema scores.
Progression of small airway abnormalities in chronic obstructive pulmonary disease
Once the pathological changes in the airways were established, the striking correlation between the progression of physiological impairment and the degree of small airway disease suggested that inflammation of the small airways made an important contribution to the functional deterioration seen in COPD, even in the presence of emphysema. Many further studies have addressed the pathological changes of the small airways in smokers and their relationship to the flow limitation found in COPD. Of special interest are studies comparing the airway changes in smokers with various degrees of emphysema and COPD with those in nonsmokers, since they not only give a perspective on the effects of smoking and emphysema but also on ageing in the airways.
In one such study, Cosio et al. 8 compared abnormalities of the small airways in smokers and nonsmokers dying accidentally. Their pulmonary function status was not known; however, the degree of both macroscopic and microscopic emphysema, assessed by means of the mean linear intercept, did not differ between smokers and nonsmokers, suggesting that in most cases the effects of cigarettes were mild. Nonetheless, abnormalities in the membranous and respiratory bronchioles of the smokers were quite apparent, with increased goblet cells, cellular inflammatory infiltrates, muscle and respiratory bronchiolitis compared with the nonsmokers. The overall mean diameter of airways <2 mm in diameter was similar in both groups, but smokers had a significantly greater proportion of bronchioles <400 µm than nonsmokers, and this proportion was closely related to the total score of airway abnormalities. Other studies have indicated that a better relationship with airflow limitation may be obtained using the proportion of very narrowed airways of 0.2 and 0.35 mm in diameter 9–11. Of special interest, this marked narrowing may also be associated with hypoxaemia and right ventricular hypertrophy 10, 12, 13.
Wright et al. 14 studied the lungs of nonsmokers, smokers and exsmokers and reported no significant differences in individual values for total pathological scores for membranous bronchioles between current and former smokers. However, respiratory bronchioles from former smokers displayed a significant decrease in goblet cell pigment, inflammation and intraluminal macrophages in comparison with those from smokers. Wright et al. 15 also found that the wall thickness of membranous and respiratory bronchioles was increased in almost all bronchiolar size ranges in smokers as compared with lifetime nonsmokers, thus indicating that smoking is associated with an increase in airway wall thickness independent of airway size and regardless of the presence or absence of emphysema. However, for the same level of function, the degree of airway abnormality could be quite variable, suggesting that other abnormalities, probably elastic recoil losses, influence the degree of flow limitation.
Hale et al. 16 extended the study of Cosio et al. 8 by studying the lungs of a further 18 patients dying with known and measured COPD. This study is of interest since it clearly shows the progression of the small airway pathological changes from nonsmoking older individuals through smokers with mild disease to smokers dying of COPD. The cellular inflammatory infiltrate, fibrosis and muscle in the airway wall increased significantly in a stepwise fashion through the three groups and, as expected from the initial study 8, the number of airways of <400 µm in diameter increased accordingly. The mean diameter of the small airways tended to decrease, but the range of diameters was so large that no significant differences could be found between even patients with the most severe COPD and nonsmokers. A similar wide range was found in all the airway inflammatory abnormalities measured in both groups of smokers, indicating that not all smokers react in the same fashion to cigarette smoke, thus suggesting that some are more prone to developing small airway abnormalities than others (fig. 2⇓). Not surprisingly, smokers dying of COPD showed more emphysema in their lungs than nonsmokers and patients with mild COPD. The degree of emphysema assessed macroscopically correlated with measurements of all abnormalities found in the small airways. With this large degree of intercorrelation, it would not be surprising if, in severe COPD, the degree of emphysema were to override correlations between morphology of the small airways and function. Nonetheless, Hale et al. 16 found that the degree of airflow limitation correlated not only with the degree of emphysema but also with the mean airway diameter and the proportion of airways of <400 µm in diameter, a function of the total pathological score of the small airways.

Scores relating to peripheral airways disease among nonsmokers (NS), smokers (S) and smokers with chronic airflow obstruction (S‐CAO): a) inflammation; b) fibrosis; c) smooth muscle; and d) goblet cell. Horizontal bars represent mean values. *: p<0.05; #: nonsignificant. (From 16).
Similar results were obtained by Nagai et al. 17 in patients dying of COPD. In their study, ante mortem flow rates correlated with the degree of macroscopic emphysema and also with the proportion of airways of <400 µm in diameter and the degree of deformity of the bronchioles. They interpreted these findings as meaning that decreases in flow were secondary to emphysema, causing loss of elastic recoil and airway obstruction caused by an excessive number of very small bronchioles and deformity of the bronchiolar lumina. It was also clear from this study that, for the same degree of airflow limitation, smokers with lesser amounts of emphysema had more diseased small airways, suggesting that the combined effect of loss of recoil secondary to emphysema and increase in airways resistance secondary to small airway abnormalities produces the airflow limitation in COPD.
The relationship between small airway diameters, a function of the proportion of airways of <400 µm in diameter in the lung, and lung disease in smokers is of interest. Smokers in the studies mentioned previously showed a significant increase in the proportions of airways of <400 µm in diameter compared with nonsmokers, and this proportion correlated with the severity of the pathological abnormalities in these airways. This correlation suggests that inflammatory and fibrotic reactions in the airways result in subtle degrees of airway narrowing, which are best detected as an increased proportion of airways of <400 µm in diameter. However, this explanation may not fully account for the relationship between disease and the calibre of the small airways. The mean bronchiolar diameter and the percentage of airways of <400 µm in diameter are highly variable even in nonsmokers 8, and, in one study, the mean bronchial diameter in young adults was found to vary over a two-fold range 18. These data suggest that small airway calibre may be constitutionally determined, secondary to diseases in childhood or both, and that airway narrowing, as measured at total lung capacity, may not be entirely due to disease. An alternative explanation is that persons with smaller airways, because of constitution or diseases in childhood, might be more susceptible to developing disease in these airways when exposed to cigarette smoke or other irritants.
The cellular inflammatory infiltrate in the airways
The earliest and constant pathological abnormality in the airway of smokers is the cellular inflammatory infiltrate throughout the wall. Inflammation, per se, may be responsible for mild airflow limitation 4, 9, 19, and it has been suggested that inflammation may lead to functional bronchiolar constriction by releasing mediators of inflammation that may act directly on bronchiolar smooth muscle 20. The chronicity of the inflammation would, in turn, produce other changes, such as fibrosis of the airway, and could increase the amount of smooth muscle either directly as a result of inflammation or indirectly as a result of chronically increased muscle tone. These changes, by increasing the thickness of the airway wall, would promote airway narrowing and airflow limitation. Finally, inflammation of the airway could play an important role in the destruction of the alveolar walls attached to the airway (alveolar attachments), and this decrease in alveolar attachments would contribute further to airflow limitation by deforming and narrowing the airway lumen.
The stimuli for this inflammatory infiltrate are not precisely known, but, very possibly, injury to the epithelium, the first structure encountered by cigarette smoke, could promote and perpetuate an inflammatory reaction in the airway. Epithelial cells have the potential to initiate airway inflammation through metabolizing arachidonic acid 21, and one product of this pathway, dihydroxyeicosatetraenoic acid is a potent signal which recruits neutrophils to the airways 22. Another system implicated in airway inflammatory responses, which could be triggered by loss or alteration of the epithelial surface, is neurogenic inflammation. Stimulation of sensory nerves in airway epithelium releases tachykinins including substance P and neurokinins A and B 23–27. Tachykinins specifically cause chemotaxis and adhesion of neutrophils in the airway circulation 28, 29, stimulate the release of inflammatory cytokines and cause degranulation of eosinophils 30, 31.
In animal studies, exposure to cigarette smoke and other irritants promotes neutrophils to appear promptly in the airways. In addition, Hulbert et al. 32 showed that, when the airway mucosa of guinea pigs is injured by inhalation of cigarette smoke, oedema occurs within 30 min, and the number of neutrophils in the airway epithelium increases five-fold from control values 6 h after injury. Not surprisingly, neutrophils are abundant in the walls of smokers' airways, and the number of submucosal neutrophils correlates significantly to the number of cigarette smoked. However, the numbers of neutrophils in the airway wall do not differ between smokers with and without airflow obstruction 33.
Other irritants also cause the airway to become inflamed. Baile et al. 34 reported that acid inhalation in dogs caused abnormalities of small airway function, which were associated with accumulation of neutrophils in noncartilaginous airways. Other research groups have documented the short- and long-term effects of other inhalational challenges. After nitrogen dioxide inhalation, rapid increases in airway resistance and airway responsiveness occur, first with an influx of neutrophils and later with mononuclear cells 35. Sulphur dioxide-induced changes have also been described 36, consisting of a marked neutrophilic infiltration of the airways and an increase in resistance. Elastase inhalation also elicits increases in bronchoalveolar lavage fluid total cell counts and is accompanied by epithelial damage, mucous plugging and neutrophil and macrophage infiltration of the bronchial mucosa 37. The effects of zone exposure have been well studied in human volunteers and probably accurately reflect the series of events occurring in the airways after repeated exposure to other respiratory irritants (e.g. NO2, SO2, cigarettes and gas oil). Acute exposure to ozone leads to an increase in the numbers of neutrophils and mononuclear cells, increases in the concentrations of total protein and interleukins (IL)‐6 and IL‐8, and a reduced concentration of glutathione in bronchoalveolar lavage fluid without changes in bronchial biopsy results. Furthermore, bronchial biopsy showed prominent neutrophilic inflammation of the airway 38.
The “early” inflammatory infiltrate found in smokers' airways, and reflected in both tests of small airway function and spirometry, probably represents the nonspecific response of the airways to injury of any type (or to certain types of inhalational insult or stimulus). Probably, the main difference between the cigarette insult and the others described above is the chronic inflammatory stimuli produced by the daily cigarette consumption. Therefore, it would seem likely that the majority of smokers will develop chronic nonspecific inflammation of the airway and probably lung parenchyma, and, for some reason, some smokers will develop severe abnormalities of the airways and emphysema that translate into clinical COPD. The remainder of the smokers without COPD would still harbour the nonspecific neutrophilic and macrophage infiltrate, but with otherwise normal airways and lung parenchyma and “mild” functional changes that never become clinical.
The overall cellular inflammatory infiltrate in smokers' airways has been described by Finkelstein et al. 39, who carefully differentiated and quantified the number of cells per cubic millimetre of airway wall in smokers', divided according to type of emphysema, and nonsmokers' lungs obtained at surgery. They found large numbers and also wide variation in the number of cells within the cases and also within the airways of the same patients (table 1⇓). Patients with centrilobular emphysema (CLE) tended to have more inflammatory cells than those with panlobular emphysema (PLE). It can be concluded from their results that the peripheral airways of cigarette smokers exhibit a large number and variety of inflammatory cells and that the extent of the cellular infiltration shows a wide variability, indicating that inflammation is unevenly distributed throughout the small airways. The wide variation in cell number has implications for studies of airway inflammation since sampling of airways using small biopsy samples may not give adequate representation of the inflammatory cell population in the small airways.
Airway wall cell densities
Subsequently, other authors phenotyped the T‐lymphocytes found in airway biopsy samples and lung specimens and characterized them as predominantly CD8+ T‐cells. Saetta et al. 40 investigated the differences in airway inflammation between smokers who develop COPD and those who do not by examining surgical specimens obtained from two groups of smokers: 1) asymptomatic smokers with normal lung function (no COPD); and 2) symptomatic smokers with abnormal lung function (COPD). Both groups were of similar age and smoking history. It was found that smokers with COPD showed evidence of airway remodelling with a measurable increase in smooth muscle mass. The differential cell counts of the cellular inflammatory infiltrates in the small airways showed that the only difference between the two groups was an increased number of CD8+ T‐cells in the airway wall of smokers with COPD as compared to the healthy smokers. All the other cell types, including neutrophils occurred in similar numbers in both groups of smokers. Interestingly, CD8+ T‐cell numbers not only were increased in these subjects but also correlated negatively with the degree of airflow limitation. Similar findings in the larger airways have been reported by O'Shaughnessy et al. 41, who demonstrated an increased number of CD8+ T‐cells in bronchial biopsy samples obtained by bronchoscopy in subjects with COPD compared to smokers without COPD.
Lams et al. 42 obtained similar results in a group of smokers and nonsmokers undergoing lung resection for lung masses. Similar to Finkelstein et al. 39, they encountered large variation in the density of inflammatory cells in general, and T‐cells in particular. However, they were able to demonstrate a significantly increased CD8+/CD3+ T‐cell ratio in smokers compared to never-smokers and exsmokers, and a significantly diminished CD4+/CD3+ T‐cell ratio in smokers. The number of neutrophils in the airway submucosa was extremely variable and did not differ between smokers and exsmokers, although it was much lower in the never-smokers. It is of interest to note that they showed a significant increase in the number of eosinophils and activated eosinophils in the airways of smokers. Unfortunately, they did not study the airway reactivity of their patients, as a relationship between bronchodilator response or airway reactivity and the number of eosinophils could have clarified the bronchodilator responses found in some smokers with COPD.
The findings of increased numbers of T‐lymphocytes, and especially CD8+ T‐cells, only in smokers who develop COPD is intriguing and supports the notion that T‐cell inflammation in the lungs is important, and may be essential for the development of COPD. In other words, only smokers, who for some reason develop a T‐lymphocyte inflammation, would develop COPD. If this were the case, an immunological reaction (mediated by CD8+ T‐cells) developing in some smokers might be the turning point from early inflammation to subclinical, and then clinical COPD. Predisposing and risk factors for the development of COPD in smokers could then be related to “immunological tolerance”, an easier phenomenon to understand and investigate.
The relationship of small airway abnormalities and emphysema in smokers
Abnormalities of the airways and how they affect airflow obstruction in COPD cannot be considered as an isolated factor in smokers. In order to understand these abnormalities, it is important to analyse the alterations in the airways vis‐à‐vis the emphysema smokers develop.
Leopold and Gough 43 showed that the small airways were usually inflamed in lungs with CLE, but, in contrast, seldom in patients with predominant PLE. Similar observations have been reported by Anderson and Foraker 44 who felt that CLE and PLE were two different diseases.
Physiological evidence suggesting that smokers could develop two different types of emphysema was provided by Eidelman et al. 45. They found that smokers with COPD had different patterns of functional abnormalities. Some exhibited pressure/volume curves typical of emphysema and resembling those seen in α1‐antitrypsin deficiency with high compliance and low elastic recoil pressure at high lung volumes. Approximately half of their subjects demonstrated low or normal compliance and, despite similar elastic recoil, lower FEV1 and pressure/volume curves not typical of emphysema.
On the basis of these findings, Kim et al. 46, reasoned that such dissimilar functional behaviour ought to correspond to different parenchymal morphological abnormalities: PLE in smokers with mechanical characteristics similar to those of α1‐antitrypsin deficiency, and CLE for the others. They tested this hypothesis in 34 patients undergoing lung resection who had undergone pulmonary function tests before surgery. Emphysema was assessed microscopically, characterized as CLE or PLE using the available definitions and quantified using the mean linear intercept. Both types of emphysema could be found in this population of smokers; 18 patients had pure or predominant CLE, and 16 had pure or predominant PLE. The mechanical properties of the two types of emphysema were found to be different. Patients with PLE demonstrated a higher compliance and a higher constant of elasticity than those with CLE. As the emphysema worsened, compliance, the elasticity constant k, and elastic recoil at 90% of total lung capacity worsened in PLE, but did not change in CLE. Moreover, losses of elasticity correlated significantly with the extent of emphysema in lungs with PLE, but not in CLE. These findings suggest that the mechanical properties of the two types of emphysema differ, and that these differences become more marked with the progression of airspace enlargement, suggesting that the two types of lung destruction are different from the start.
Another important difference between the two types of emphysema was the extent of the airway abnormalities. Lungs with CLE yielded higher total pathological scores of the small airways than those with PLE, mainly ascribable to increased muscle and fibrosis of the airway wall, and as a result of the more severe pathological involvement, lungs with CLE had more airways of <400 µm in diameter 47 and smaller mean airway diameters 42. There was also a definite tendency for small airways in the upper lung to be more diseased than those in the lower lung, a topographical distribution that corresponds to CLE 8. It is also known that disease in the small airways temporally precedes CLE 4, and that emphysema lesions first appear in the respiratory bronchiole, where the inflammatory reaction, particularly macrophage accumulation, is most intense 3, 4. The present authors believe that these various observations are complementary and support the hypothesis that small airway disease is causally related to the development of CLE.
Another important difference in the behaviour of the airways in the two emphysemas is the degree of airway reactivity, a phenomenon frequently, but not consistently found in COPD. Finkelstein et al. 39 carefully analysed the airway dimensions and inflammatory infiltrate in the lungs of smokers and nonsmokers and found that airway reactivity was significantly higher (and abnormal) in smokers with CLE than in smokers with PLE. Both groups of smokers had a similar smoking history, age and FEV1, although, in smokers with CLE, the small airways were narrower, thicker and more deformed. The degree of airway reactivity correlated with the thickness of the internal wall and the numbers of T‐lymphocytes in the airway wall in smokers with CLE, but not in those with PLE (fig. 3⇓). In a larger number of smokers with CLE and PLE and across a broader range of FEV1, Cosio et al. 47 showed by multiple regression analysis that airway reactivity in CLE is determined by the degree of pathological abnormality (cellular inflammation and amount of muscle and fibrosis) in the small airways, whereas, in PLE, it is determined by the FEV1 47. These observations indicate that there is a clear difference in responsiveness between the emphysema types, suggesting that responsiveness is not just a reaction to smoking, but either a reaction developing in some smokers or an abnormality initially present in some smokers, which in combination with exposure to cigarettes, determines the development of a type of lung disease with severe remodelling of the airways, i.e. CLE.

Provocative concentration of methacholine causing a 20% fall in forced expiratory volume in one second (PC20) versus airway wall T‐lymphocyte density in nonsmokers (▾) and patients with centrilobular (CLE, •) and panlobular emphysema (PLE, ○). There was significant negative correlation in CLE (——, slope=−1×10−4, r=0.50, p<0.05), but no relationship in PLE (------, slope=1×10−7, r=0.05, p=ns). (From 39).
Not surprisingly, the pathophysiology of flow limitation in smokers, a function of airway resistance and elastic recoil pressures, differs between the two types of emphysema. Flow was shown to decrease as airway abnormalities increased in CLE, but no relationship between flow and airway disease could be found in PLE. In contrast, flow decreases significantly as elasticity decreases in PLE, but not in CLE 46. Some patients with CLE exhibit decreased flow with normal or even increased elastic recoil pressures whereas, in those with PLE flow limitation is always accompanied by loss of recoil. These findings clarify the pathogenesis of airflow limitation in smokers, indicating that, in CLE, loss of flow is primarily a function of airway abnormalities, with elastic recoil loss playing an additive role. By contrast, in patients with PLE, the flow limitation seems to be mainly a function of reduced elastic recoil; additional airway abnormalities could worsen flows even further in these cases.
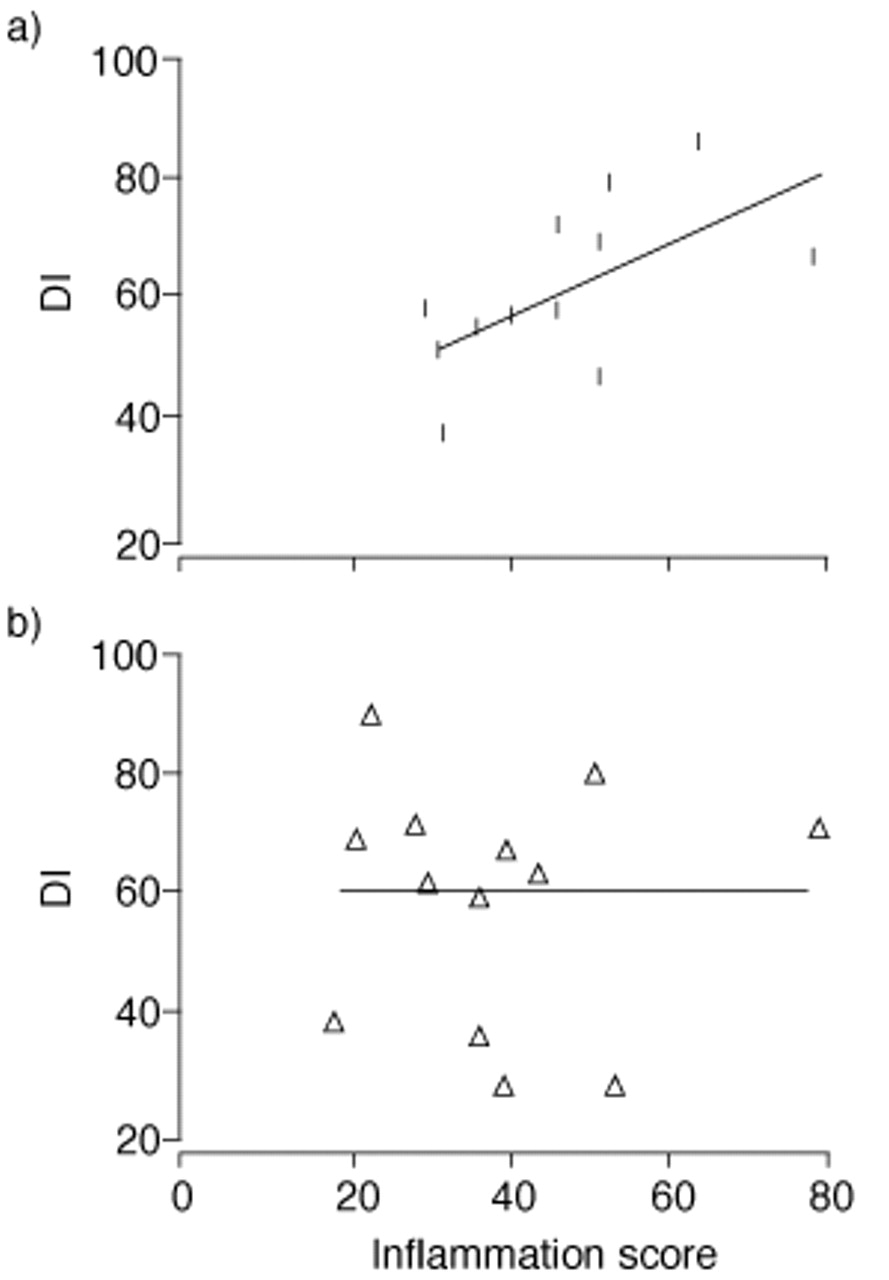
These studies are of interest because they not only clarify the mechanisms of airflow limitation and the role of the bronchioles in COPD, but they also confirm the possibility of smokers developing two diseases with different pathogenetic mechanisms. The diffuse destruction seen in PLE might result from a blood-borne mechanism. Conversely, the uneven pattern of lung destruction seen in CLE is associated with more severe abnormalities of the small airways, suggesting that centrilobular destruction is related to airborne factors and intimately related to the airway inflammatory process. The findings of Saetta et al. 48, who investigated the relationship between the inflammation of the small airways and the extent of parenchymal destruction using the destructive index 49 in the lungs of smokers with CLE and with PLE, are in agreement with this hypothesis. In lungs with CLE, a close correlation between inflammation of the airways and parenchymal destruction was evident. This correlation was not present in lungs with PLE (fig. 4⇓). Thus, it is likely that the inflammatory reaction seen in and around small airways and respiratory bronchioles spreads centrifugally to the parenchyma surrounding these airways and eventually destroys the alveolar walls attached to the airways, respiratory bronchioles and alveolar ducts. The preservation of alveolar structure and size, with the concomitant destruction of alveolar ducts and respiratory bronchioles seen in CLE, supports this possibility.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Correlation between the inflammation score in the small airways and the destructive index (DI) in: a) centrilobular emphysema (CLE); and b) panlobular emphysema (PLE). Significant correlation occurs in CLE (r=0.617, p<0.025), but not in PLE (r=−0.009, p=ns). (From 48).
These studies provide a new basis for the investigation of lung disease in smokers. If the intimal pathogenetic mechanisms of the two types of emphysema in smokers are different, as the evidence suggests, the study of cigarette-induced lung disease as a single entity will further delay understanding of chronic obstructive pulmonary disease.
- Received April 6, 2001.
- Accepted April 17, 2001.
- © ERS Journals Ltd
References









