





Abstract
Increased synthesis of endothelin-1 (ET-1) by human vascular endothelial cells (HVECs) in response to hypoxia underscores persistent vasoconstriction observed in patients with pulmonary hypertension. The molecular mechanism whereby hypoxia stimulates ET-1 gene transcription is not well understood. Here we report that megakaryocytic leukemia 1 (MKL1) potentiated hypoxia-induced ET-1 transactivation in HVECs. Disruption of MKL1 activity by either a dominant negative mutant or small interfering RNA mediated knockdown dampened ET-1 synthesis. MKL1 was recruited to the proximal ET-1 promoter region (−81/+150) in HVECs challenged with hypoxic stress by the sequence-specific transcription factor serum response factor (SRF). Depletion of SRF blocked MKL1 recruitment and blunted ET-1 transactivation by hypoxia. Chromatin immunoprecipitation analysis of the ET-1 promoter revealed that MKL1 loss-of-function erased histone modifications consistent with transcriptional activation. In addition, MKL1 was indispensable for the occupancy of Brg1 and Brm, key components of the chromatin remodeling complex, on the ET-1 promoter. Brg1 and Brm modulated ET-1 transactivation by impacting histone modifications. In conclusion, our data have delineated a MKL1-centered complex that links epigenetic maneuverings to ET-1 transactivation in HVECs under hypoxic conditions.
INTRODUCTION
Rhythmic vascular tone is essential for the internal homeostasis. Vascular tone is maintained by the regular constriction and relaxation of vascular smooth muscle cells under the influence of a group of humoral factors collectively referred to as vasoactive substances (1). Endothelin-1 (ET-1) is by far the most potent endothelium-derived vasoconstrictor. Elevated ET-1 production is synonymous with persistent vessel constriction and disrupts normal vascular tone, contributing to a range of diseased states including pulmonary hypertension, atherosclerosis and cardiac fibrosis (2–4).
ET-1 levels are tightly regulated primarily at the transcriptional level under various pathophysiologic conditions. A plethora of pro-inflammatory mediators, including LPS and tumor necrosis factor alpha (TNF-α), substantially accelerate ET-1 transcription via NF-κB as means of exacerbating vascular injury (5,6). Reactive oxygen species and shear stress, two major culprits of cardiovascular pathology, stimulate ET-1 transcription by sharing a similar MAP kinase signaling pathway (7,8). Despite the numerous sequence-specific transcription factors and signaling cascades uncovered so far for the ET-1 gene, it remains largely unanswered how the transcriptional status of the ET-1 gene correlates with its chromatin structure.
Megakaryocytic leukemia 1 (MKL1), variously termed myocardin-related transcription factor A (MRTF-A) or myeloid acute leukemia (MAL), is a transcriptional regulator initially identified as a cofactor for serum response factor (SRF). MKL1 potentiates SRF-dependent transactivation of genes specific to cells of the muscle lineage thereby playing a key role in the phenotypic modulation of smooth muscle cells (9,10). Unlike its close sibling myocardin whose expression is restricted to smooth and cardiac muscles, MKL1 is universally present in all cell types within the vasculature, indicative of a more systemic role for MKL1 in cardiovascular disease. Indeed, a number of investigations have painted MKL1 as crucial in the pathogenesis of myocardial infarction, cardiac hypertrophy and atherosclerosis, attributable to its role in fibroblast, myocyte and endothelial cells, respectively (11–13). Of intrigue, MKL1 seems to be able to engage multiple components of the epigenetic machinery in the transcription of its target genes (13–17).
Recent investigation in our laboratory harnessing lentivirus to deliver short-hairpin RNA (shRNA) targeting MKL1 revealed that MKL1 depletion ameliorated hypoxia-induced pulmonary hypertension (HPH) in rats (Z. B. Yuan and Y. Xu, manuscript under review elsewhere). Because ET-1 overproduction as a result of transcriptional activation by hypoxic stress is believed to be a central theme of HPH (18,19), we hypothesized that MKL1 might directly regulate ET-1 transactivation in endothelial cells in response to hypoxia. Our data as summarized here portrays MKL1 as an epigenetic orchestrator that connects chromatin modification to hypoxia-induced ET-1 transcription.
MATERIALS AND METHODS
Cell culture
Human vascular endothelial cells (HVECs/EAhy926, ATCC), human adenoma cells (SW-13, ATCC) and human embryonic kidney cells (HEK293, Invitrogen) were maintained in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Hyclone). Murine embryonic fibroblast cells were isolated from wild type or MKL1 knockout C57/BL6 mice (20). Where indicated, hypoxia (1% O2) was achieved by a mixture of ultra-high purity gases (5% CO2, 10% H2, 85% N2) in a 37°C incubator (Thermo Fisher).
Plasmids, transient transfection and luciferase assay
Details for DNA constructs used in this study can be found in Supplementary Material. Small interfering RNA (siRNA) for MKL1, SRF, Brg1 and Brm have been described (13,21); validation of all siRNAs used in this study can be found in Supplementary Figure S5. Transient transfections were performed with Lipofectamine 2000 (Invitrogen) essentially as previously described (22). Luciferase activities were assayed 24–48 h after transfection using a luciferase reporter assay system (Promega).
Animals
All animal experiment protocols were approved by the Committee on Ethical Practice of Animal Studies of the Third Military Medical University. Details can be found in Supplementary Material.
RNA isolation and real-time polymerase chain reaction
RNA was extracted with the RNeasy RNA isolation kit (Qiagen). Reverse transcriptase reactions were performed using a SuperScript First-strand Synthesis System (Invitrogen). Real-time polymerase chain reaction (PCR) reactions were performed on an ABI Prism 7500 system. Primers and Taqman probes were purchased from Applied Biosystems.
Protein extraction and western blot
Whole cell lysates were obtained by resuspending cell pellets in RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100) with freshly added protease inhibitor tablet (Roche). Nuclear proteins were prepared with the NE-PER Kit (Pierce) following manufacturer’s recommendation. Western blot analyses were performed with anti-β-actin (Sigma), anti-α-tubulin (Millipore) and anti-MKL1 (Santa Cruz SC-32909, 1:2000 dilution) antibodies.
Enzyme-linked immune absorbance assay
Enzyme-linked immune absorbance assay (ELISA) was performed using supernatant collected from cultured HVECs or rat pulmonary artery homogenates according to manufacturer’s instructions (USCN Life Sciences).
Immunofluorescence microscopy
Endothelial cells were fixed with 4% formaldehyde, permeabilized with TBST, blocked with 5% bovine serum albumin and incubated with indicated primary antibodies overnight. After several washes with phosphate buffered saline, cells were incubated with FITC-labeled secondary antibodies (Jackson) for 30 min. DAPI (Sigma) was added and incubated with cells for 5 min before observation. Immunofluorescence was visualized on a co-focal microscope (LSM 710, Zeiss).
Chromatin immunoprecipitation and Re-ChIP
Chromatin Immunoprecipitation (ChIP) and Re-ChIP assays were performed essentially as described before (13). Aliquots of lysates containing 200 μg of protein were used for each immunoprecipitation reaction with anti-MKL1, anti-SRF, anti-Brg1, anti-Brm (Santa Cruz), anti-acetyl histone H3 and H4, anti-monomethyl, anti-dimethyl and anti-trimethyl H3K4, anti-trimethyl H3K9 (Millipore) or pre-immune IgG. Precipitated genomic DNA was amplified by real-time PCR with primers listed in Supplemental Table S1.
Statistical analysis
One-way analysis of variance with post hoc Scheffe analyses were performed using an SPSS package. P values < 0.05 were considered statistically significant and designated*.
RESULTS
MKL1 activates ET-1 transcription in response to hypoxia in endothelial cells
Our recent finding has implicated MKL1 in the pathogenesis of hypoxia pulmonary hypertension. Because upregulation of ET-1 transcription by hypoxia represents a key step in HPH, we sought to determine whether MKL1 could be involved in this process. As shown in Figure 1A, MKL1 dose-dependently activated an ET-1 promoter construct in HVECs. More importantly, MKL1 markedly potentiated transactivation of ET-1 promoter under hypoxic conditions (Figure 1B). In accordance, endogenous ET-1 levels were upregulated by MKL1 in response to hypoxic challenge (Figure 1C). Activities of the class II transactivator (CIITA) promoter, on the other hand, were not altered by MKL1 under either normoxic or hypoxic conditions (Supplementary Figure S1A).
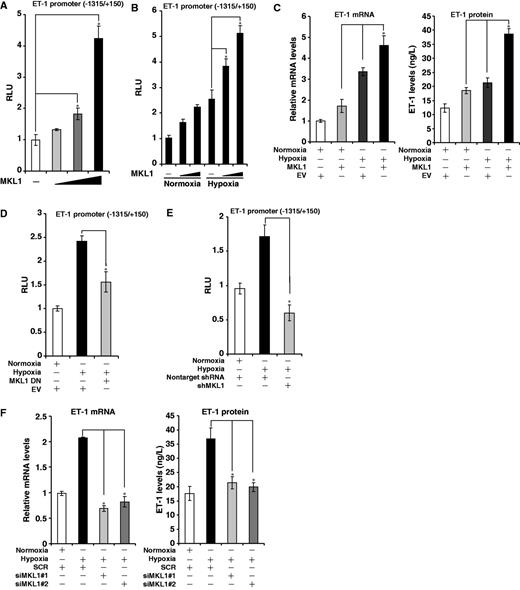
MKL1 activates ET-1 transcription in response to hypoxia in endothelial cells. (A) A rat ET-1 promoter-luciferase construct was transfected into HVECs with increasing amount of MKL1. Data are expressed as relative luciferase unit (RLU). (B) A rat ET-1 promoter-luciferase construct was transfected into HVECs with MKL1 followed by exposure to 1% O2. Data are expressed as RLU. (C) An empty vector (EV) or MKL1 expression construct was transfected into HVEC followed by exposure to 1% O2. ET-1 levels were measured by qPCR and ELISA. (D) A rat ET-1 promoter-luciferase construct was transfected into HVECs with EV or DN MKL1 followed by exposure to 1% O2. Data are expressed as RLU. (E) A rat ET-1 promoter-luciferase construct was transfected into HVECs with shRNA plasmid targeting MKL1 or control shRNA (non-target) followed by exposure to 1% O2. Data are expressed as RLU. (F) siRNA targeting MKL1 (siMKL1) or scrambled RNA (SCR) was transfected into HVECs followed by exposure to 1% O2. ET-1 levels were measured by qPCR and ELISA.
Meanwhile disruption of MKL1 activity by expression of either a dominant negative (DN) construct (Figure 1D) or short hairpin RNA targeting MKL1 (shMKL1, Figure 1E) abrogated ET-1 promoter transactivation by hypoxia. These data were corroborated by the observation that ET-1 promoter activity was significantly downregulated and failed to response to hypoxic challenge in mouse embryonic cells lacking MKL1 (Supplementary Figure S1B). In addition, two separate pairs of siRNA targeting MKL1 both abolished the stimulation of endogenous ET-1 synthesis by hypoxia (Figure 1F). Finally, when we injected lentivirus carrying shMKL1 into rats that were then allowed to develop HPH, induction of ET-1 protein in pulmonary arteries was decreased compared with the control group (Supplementary Figure S1C).
To pin down the binding site for MKL1 on the ET-1 promoter, we transfected ET-1 promoter constructs with serial deletions with or without MKL1 into cells. Interestingly, MKL1 was able to activate the minimal ET-1 promoter (−81/+150, Figure 2A). In agreement, shMKL1-mediated ET-1 transrepression was effective on the smallest promoter (Figure 2B). Finally, we performed ChIP assays to examine whether MKL1 indeed could bind to this region of the ET-1 promoter. Indeed, occupancy of MKL1 on the proximal promoter was increased with time under hypoxic conditions (Figure 2C). Together, our results indicate that MKL1 is both sufficient and necessary for hypoxia-induced transactivation of the ET-1 gene.
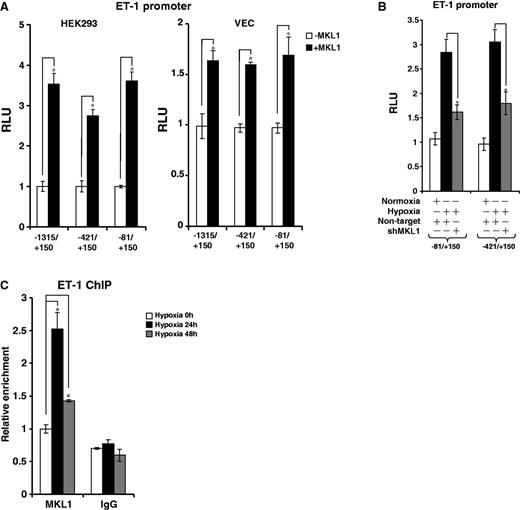
Hypoxia stimulates the recruitment of MKL1 to the ET-1 promoter. (A) HEK293 and endothelial cells were transfected with ET-1 promoter-luciferase constructs of different lengths with or without MKL1. Data are expressed as RLU. (B) HEK293 cells were transfected with ET-1 promoter-luciferase constructs of different lengths with shMKL1 or non-targtet shRNA. Data are expressed as RLU. (C) HVECs were exposed to 1% O2 and harvested at indicated time points. ChIP assays were performed with anti-MKL1 or IgG.
MKL1-mediated ET-1 transactivation relies on SRF
Close examination of this region (−81/+150) revealed a couple of non-classical CArG sites within this region (Supplementary Figure S2A), indicating that SRF might potentially play a role in MKL1-dependent ET-1 transcription in response to hypoxia. Several lines of evidence support this hypothesis. First, interference with endogenous SRF activity by either DN mutant (Supplementary Figure S2B) or siRNA (Figure 3A, Supplementary Figure S2C) completely blocked the activation of the minimal ET-1 promoter by hypoxia. Second, hypoxia stimulated the recruitment of SRF to the ET-1 promoter in both ChIP assay (Figure 3B) and gel shift assay (Supplementary Figure S2G). Importantly, depletion of SRF prevented the binding of MKL1 (Figure 3C), while re-ChIP assay confirmed the existence of an SRF-MKL1 complex on this region in hypoxia-treated HVECs (Supplementary Figure S2D), reinforcing the notion that SRF may be responsible for bringing MKL1 to the ET-1 promoter. In accordance, MKL1 failed to activate the ET-1 promoter in the absence of SRF (Figure 3D and E). We also mutated the putative CArG box within the ET-1 reporter; neither MKL1 overexpression nor hypoxia was able to activate the mutant construct (Supplementary Figure S2E and F). Finally, SRF knockdown markedly attenuated ET-1 synthesis and release from HVECs under hypoxic conditions (Figure 3F). Collectively, these data demonstrate that SRF is involved in hypoxia-induced ET-1 transactivation bridging MKL1 and the target chromatin.
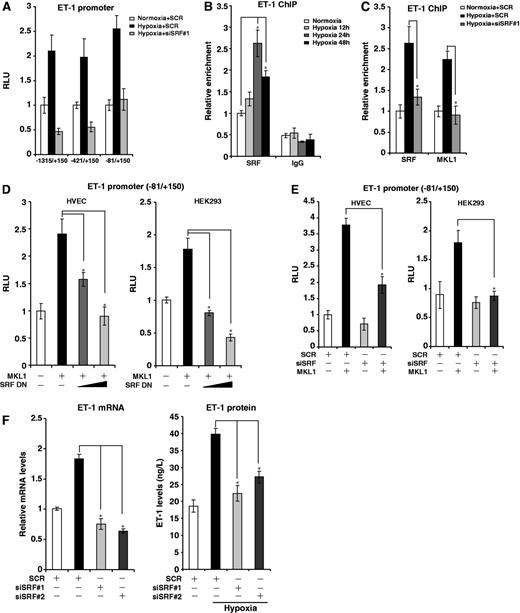
MKL1-mediated ET-1 transactivation relies on SRF. (A) HVECs were transfected with ET-1 promoter-luciferase constructs of different lengths and indicated siRNA followed by exposure to 1% O2. Data are expressed as RLU. (B) HVECs were exposed to 1% O2 and harvested at indicated time points. ChIP assays were performed with anti-SRF or IgG. (C) HVECs were transfected with indicated siRNAs followed by exposure to 1% O2. ChIP assays were performed with indicated antibodies. (D) A rat ET-1 promoter-luciferase construct was transfected into HVECs and HEK293 cells with MKL1 and DN SRF. Data are expressed as RLU. (E) A rat ET-1 promoter-luciferase construct was transfected into HVECs and HEK293 cells with MKL1 and indicated siRNA. Data are expressed as RLU. (F) HVECs were transfected with indicated siRNAs followed by exposure to 1% O2. ET-1 mRNA and protein levels were measured by qPCR and ELISA.
MKL1 coordinates key epigenetic alterations on the ET-1 promoter
Hypoxia-induced transactivation is associated with dynamic histone modifications surrounding the promoter region of genes. Because several recent reports have suggested a potential connection between MKL1 and the epigenetic machinery, we examined whether MKL1 could influence these alterations. As shown in Figure 3, depletion of MKL1 by siRNA (Figure 4A) significantly decreased the accumulation of acetylated histone H3 (Figure 4B), di- and trimethylated histone H3K4 (Figure 4E and F), all of which herald transcriptional activation, on the proximal ET-1 promoter in HVECs under hypoxic stress. On the other hand, MKL1 elimination also restored the levels of trimethylated histone H3K9, a prominent marker for transcriptional repression (Figure 4G). By comparison, although there was an increase in the levels of acetylated histone H4 on the ET-1 promoter, MKL1 had little impact on this modification (Figure 4C). Nor did MKL1 affect monomethylated H3K4 (Figure 4D). Therefore, MKL1 modulates ET-1 transactivation likely by selectively manipulating histone modifications.
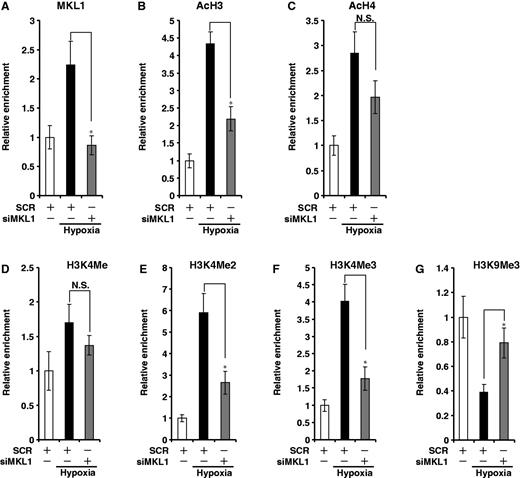
MKL1 coordinates key epigenetic alterations on the ET-1 promoter. (A–G) HVECs were transfected with siMKL1 or SCR followed by exposure to 1% O2. ChIP assays were performed with anti-MKL1 (A), acetyl histone H3 (B) and H4 (C), methylated histone H3K4 (D, E, F) and H3K9 (G).
MKL1 recruits Brg1 and Brm to activate ET-1 transcription
Previous investigations have shown that the chromatin remodeling proteins Brg1 and Brm are required for MKL1-dependent transcription of smooth muscle-specific genes. We hypothesized that Brg1/Brm might be indispensible for MKL1-mediated ET-1 transactivation. Indeed, ChIP assay demonstrated that there was increased occupancy of Brg1 and Brm on the proximal ET-1 promoter when HVECs were cultured in 1% O2 (Figure 5A). Of note, an MKL1-Brg1/Brm complex could be established on the ET-1 promoter in response to hypoxia (Figure 5B). On the contrary, recruitment of Brg1 and Brm was blocked in the absence of MKL1 (Figure 5C).
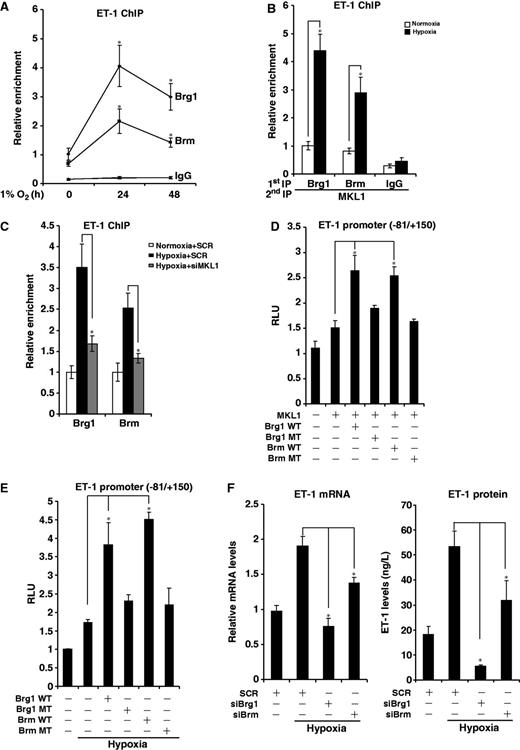
MKL1 recruits Brg1 and Brm to activate ET-1 transcription. (A) HVECs were exposed to 1% O2 and harvested at indicated time points. ChIP assays were performed with indicated antibodies. (B) HVECs were exposed to 1% O2 for 24 h. Re-ChIP assay was performed with indicated antibodies. (C) HVECs were transfected with siMKL1 or SCR followed by exposure to 1% O2. ChIP assays were performed with anti-Brg1 or Brm. (D) A rat ET-1 promoter-luciferase construct was transfected into HVECs with expression constructs for MKL1, Brg1 or Brm as indicated. Data are expressed as RLU. (E) A rat ET-1 promoter-luciferase construct was transfected into HVECs with expression constructs for Brg1 or Brm followed by exposure to 1% O2. Data are expressed as RLU. (F) HVECs were transfected indicated siRNA followed by exposure to 1% O2. ET-1 mRNA and protein levels were measured by qPCR and ELISA.
To directly examine the involvement of Brg1/Brm in ET-1 transcription, the following experiments were performed. Wild type, but not enzyme dead, Brg1 and Brm enhanced MKL1-induced transactivation of the ET-1 promoter in HVECs (Figure 5D) and SW-13 cells that lack endogenous Brg1 and Brm (Supplementary Figure S3A). Similarly, Brg1 and Brm also augmented the ET-1 promoter activity in HVECs (Figure 5E) and SW-13 cells (Supplementary Figure S3B) under hypoxic conditions. In contrast, siRNA-mediated depletion of Brg1/Brm abrogated MKL1/hypoxia-induced ET-1 transactivation in HVECs (Supplementary Figure S3C and D). Finally, Brg1/Brm knockdown decreased ET-1 synthesis and release from HVECs in response to hypoxia (Figure 4F). Together, these data suggest that Brg1 and Brm are indispensable for hypoxia-induced and MKL1-mediated transactivation of the ET-1 gene.
Brg1/Brm orchestrate key histone modifications on the ET-1 promoter
Brg1 and Brm are known to forge dialogues with other branches of the epigenetic machinery, so we asked whether Brg1 and Brm could directly impact the histone modifications surrounding the ET-1 promoter. As shown in Figure 6, knockdown of Brg1 or Brm prevented the dimethylation and trimethylation of histone H3K4 as well as acetylation of histone H3 on the ET-1 promoter. On the contrary, histone H4 acetylation and H3K4 monomethylation remained unaltered. These patterns are highly reminiscent of the chromatin structure surrounding the ET-1 promoter in MKL1-depleted cells. Therefore, we propose that MKL1, likely through recruiting and interacting with Brg1/Brm, shape the chromatin structure of the ET-1 gene to facilitate transcription in response to hypoxia in HVECs.
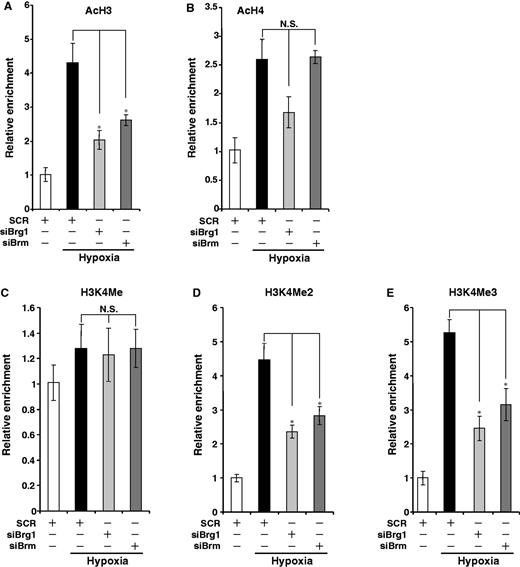
Brg1/Brm orchestrate key epigenetic alterations on the ET-1 promoter. (A–E) HVECs were transfected with siMKL1 or SCR followed by exposure to 1% O2. ChIP assays were performed with indicated antibodies.
Hypoxia activates MKL1 in endothelial cells
Finally, we tackled the question whether hypoxia could directly impact the expression and/or activity of MKL1. One percent O2 significantly upregulated mRNA and protein levels of MKL1 in HVECs peaking at 24 h following exposure to hypoxia and remaining elevated as late as 72 h after treatment (Figure 7A and B). More importantly, MKL1 expression levels were increased in pulmonary arteries in vivo in rats that were housed in a hypoxic chamber for 28 days (Figure 7C). Expression of MKL2, a closely related family member, was also activated by hypoxia (Supplementary Figure S4B) although MKL2 failed to activate ET-1 transcription in reporter assay (Supplementary Figure S4C). The induction of MKL1 expression can be attributed to, at least in part, increased transcription rate as hypoxia up-regulated luciferase activity driven by the MKL1 promoter (Figure 7D). MKL1 activity is known to be regulated by subcellular localization. Therefore, we evaluated the effect of hypoxia on MKL1 compartmentation. Using both cytoplasmic/nuclear protein fractionation (Figure 7E) and co-focal immunofluorescence microscopy (Figure 7F, Supplementary Figure S4A), we were able to show that there was increased nuclear accumulation of MKL1 in response to 1% O2 in endothelial cells. Collectively, these data illustrate that hypoxia activates MKL1 in endothelial cells both in vitro and in vivo.
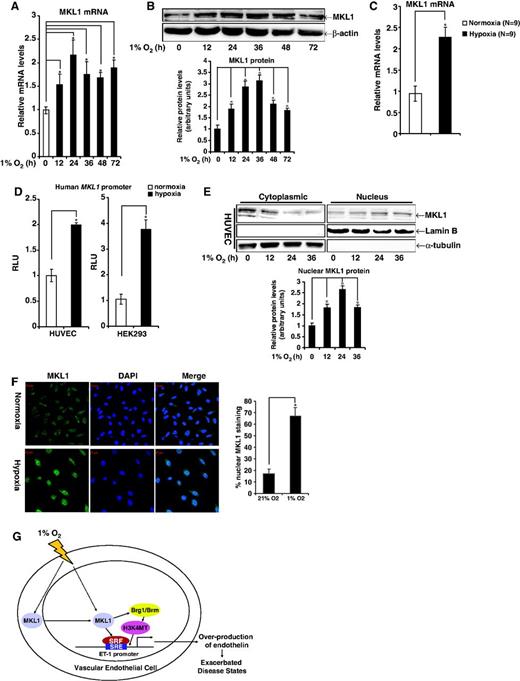
Hypoxia activates MKL1 in endothelial cells. (A and B) HVECs were exposed to 1% O2. Cells were harvested at various time points after treatment and probed for mRNA (A) and protein (B) levels of MKL1 using qPCR and Western, respectively. (C) SD rats were housed under 1% O2 for 28 days before sacrifice. mRNA levels of MKL1 in pulmonary arteries were probed by qPCR. (D) A luciferase construct driven by the human MKL1 promoter (pGL3-MKL1) was transfected into HVECs and HEK293 cells followed by exposure to 1% O2. Luciferase activities were corrected for protein concentration and transfection efficiency. Data are expressed as RLU. (E) HVECs were exposed to 1% O2. Cells were harvested at various time points after treatment, fractionated and probed for MRTF-A by Western. (F) HVECs were exposed to 1% O2 for 48 h. Cellular MKL1 was visualized by immunofluorescence staining. The nuclei were counterstained with DAPI. Scale bar: 20 μM. Nuclear MKL1 staining was quantified by Image J and expressed as percentage of overall MKL1 staining. (G) A schematic model depicting hypoxia activates MKL1, which in turn mediates ET-1 transactivation in endothelial cells.
DISCUSSION
As highlighted in several recent reports, MKL1 is emerging as a key regulator of cardiovascular disease (11–13). The present study investigates the role of MKL1 in hypoxia-induced ET-1 transcription in HVECs, a key process involved in pulmonary hypertension. Our data allude to a scenario in which MKL1 functions as a critical node in the epigenetic machinery linking chromatin alterations to the induction of ET-1 expression.
It has been well documented that MKL1 regulates transcription by engaging the epigenetic machinery (13–17). Our data indicate that MKL1 coordinates histone H3K4 di- and trimethylation on the ET-1 promoter. On the other hand, whereas MKL1 seems to be crucial for H3 acetylation, it is dispensable for H4 acetylation on the ET-1 promoter. ET-1 transcription is known to exhibit varied dependency on histone acetylation. Pro-inflammatory stimuli (TNF-α plus IFN-γ) induced ET-1 transcription requires NF-κB–mediated histone H4 acetylation (23), whereas high glucose activates ET-1 transcription by H3 acetylation (24). Hanna et al have shown that MKL1 occupancy on the pro-angiogenic gene CCN1/CYR61 correlates with acetylation of both H3 and H4 in bladder smooth muscle cells (14). Interestingly, our previous investigation has unveiled that MKL1 is required for both H3 and H4 acetylation on the ICAM-1 promoter in response to oxLDL treatment (13). These observations collectively argue for a model wherein MKL1 converses with different components of the epigenetic machinery to regulate transcription in a cell- and context-specific manner. Recently, Xia et al have demonstrated that there is a global change in histone modifications in deoxygenated HepG2 cells, suggesting that it constitutes a common theme for hypoxia-induced gene expression (25). It will be of great interest to determine how MKL1, on a genome-wide scale, contributes to hypoxia-induced gene expression by eliciting the epigenetic machinery.
A major finding of the current investigation is that chromatin remodeling proteins Brg1 and Brm are intimately involved in hypoxia-induced ET-1 transactivation. The effect of MKL1 seems to be reliant, at least in part, on Brg1 and Brm given the observations that depletion of either Brg1 or Brm completely abolished MKL1-stimulated ET-1 activation and rendered a chromatin structure similar to the MKL1-deficient state. This is consistent with the report that Brg1 and Brm are absolutely required for MKL1-induced expression of SMC-specific genes in non-muscle cells (16,17). Mounting evidence has accumulated for a central role of Brg1 and Brm in the vasculature both during organogenesis and in diseased states. In agreement with the current data, a majority of these reports seem to indicate Brg1 as a factor accountable for decompensatory responses including pathologic cardiac hypertrophy and inflammation (26–28). Our unpublished results suggest that endothelial-specific knockdown of Brg1/Brm in mice prevents high-fat diet-induced atherosclerosis and attenuates low oxygen tension-induced pulmonary hypertension (F. Fang, D. W. Chen and Y. Xu, manuscript under consideration elsewhere), affirming the notion that hyperactive Brg1/Brm in HVECs largely dictates detrimental processes and may herald a poorer outcome.
MKL1 was initially identified as a cofactor for SRF, and to date an overwhelming number of MKL1 target genes have been confirmed to contain the CArG element within their promoter regions supporting a direct involvement of SRF. Here, we demonstrate that SRF binds to the proximal ET-1 promoter, recruits MKL1 to the same region and is indispensable for ET-1 gene transactivation in response to hypoxia. There appears to be interplay between SRF and ET-1 in the vasculature. On the one hand, SRF has been shown to mediate the harmful effects of ET-1 by stimulating the transcription of ANF and BNP (12,29). On the other hand, ET-1 is able to enhance SRF-dependent transcription and directly augment SRF expression (30,31). It is conceivable that in hypoxia-challenged HVECs ET-1, once activated by SRF, would feed to the increase of SRF levels and generate more ET-1, thereby creating a forward feedback loop that eventually cause an outpour of ET-1 into the circulation and promote the pathogenesis of HPH.
Our data show that hypoxic stress can enhance the activity of MKL1 by at least two mechanisms. First, hypoxia directly activated the transcription rate of MKL1 (Figure 7D). A survey of the MKL1 promoter region (−1585/+81) revealed several potential binding sites for transcriptional factors including HIF, NF-κB and Sp1 (Y. Y. Yang and Y. Xu, unpublished observation). Therefore, it is possible that these factors, acting alone or in combination, could be recruited to the MKL1 promoter and contribute to hypoxia-induced MKL1 transactivation. Second, hypoxia promoted the nuclear enrichment of MKL1 (Figure 7E and F). Shuttling of MKL1 between the cytoplasm and the nucleus relies on the re-organization of cytoskeletons induced by Rho family of small G proteins (32). RhoA (33), RhoB (34) and Rac (35), have all been shown to be involved in hypoxia-invoked changes in cytoskeletal structure in endothelial cells. It would be of great interest to determine the specific Rho protein(s) connecting hypoxia to MKL1 nuclear accumulation and consequently ET-1 transactivation.
In summary, a transcriptional complex that contains MKL1, SRF, Brg1/Brm and possibly other epigenetic factors (histone methyltransferase/HMT and acetyltransferase/HAT, for example) dwells on the proximal ET-1 promoter in HVECs and activates ET-1 transcription in response to hypoxia by conferring a transcriptionally friendly chromatin structure. Targeting this MKL1-centered complex may yield novel interventional solutions for HPH.
FUNDING
National Basic Science Research Program of China 973 [2012CB518201, 2012CB517503]; the Program for New Century Excellent Talents in University of China [NCET-11-0991]; Natural Science Foundation of China [31270805, 81100041]; Natural Science Foundation of Jiangsu Province [BK2012043]; the Ministry of Education [212059, 20122323411008]; the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD); the Postdoctoral Research Grant of China [20100471771] (in part). Funding for open access charge: PAPD.
Conflict of interest statement. None declared.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first three authors should be regarded as joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments