




Abstract
The Birt-Hogg-Dubé (BHD) syndrome is an inherited genodermatosis characterized by a predisposition to hamartomatous skin lesions, pulmonary cysts, and renal carcinoma. Seventy-seven renal tumors from 12 patients with germline BHD mutations were examined by DNA sequencing to identify somatic mutations in the second copy of BHD. Sequence alterations were detected in the majority of renal tumors (41 of 77, 53%), with loss of heterozygosity at the BHD locus in a minority of additional tumors (14 of 77, 17%). The somatic mutations were distributed across the entire gene, and the majority resulted in frameshifts that are predicted to truncate the BHD protein. These results support a role for BHD as a tumor suppressor gene that predisposes to the development of renal tumors when both copies are inactivated.
A small percentage of renal cell carcinomas (RCCs), which are subclassified by histology into clear-cell (75% of cases), papillary (10%–15%), and chromophobe (5%) RCCs and renal oncocytoma (3%–5%), are due to inherited cancer syndromes ( 1 , 2 ) . Each inherited cancer syndrome, such as von Hippel-Lindau (VHL), hereditary papillary cell carcinoma (HPRC), and hereditary leiomyomatosis and renal cell carcinoma (HLRCC), is characterized by the development of specific histologic types of renal cancer ( 3 , 4 ) . For example, affected members of families with VHL syndrome frequently develop clear-cell RCCs, whereas patients with HPRC are predisposed to develop type 1 papillary renal carcinomas ( 5 , 6 ) . Patients with HLRCC, by contrast, develop aggressive papillary type 2 renal carcinomas ( 7 – 9 ) .
Recently, individuals with Birt-Hogg-Dubé syndrome (BHD), a genodermatosis characterized by fibrofolliculomas (hamartomas of the hair follicle) ( 10 ) and pulmonary cysts ( 11 , 12 ) , were found to have a seven-fold higher risk over the general population of developing kidney neoplasms ( 13 ) . Unlike renal tumors in patients with other inherited kidney cancer syndromes, renal tumors from BHD patients exhibit a spectrum of histologic types, including chromophobe (34%), oncocytoma (5%), clear cell (9%), papillary (2%), and an oncocytic hybrid (50%) with features of chromophobe RCC and renal oncocytoma ( 14 – 16 ) . Germline mutations have been identified in a novel gene, BHD, in affected family members ( 17 ) . BHD encodes a protein, folliculin, which is named for the hallmark dermatologic lesions found in BHD patients. All germline mutations identified to date are frameshift or nonsense mutations that are predicted to truncate folliculin, including insertions or deletions of a tract of eight cytosines (C8) in exon 11 ( 17 , 18 ) . Mutations in this “hot spot” are found in the germline of 44% of BHD patients ( 17 ) . The high frequency of germline-inactivating mutations suggests that BHD may act as a tumor suppressor gene. Identifying a somatic “second hit” in the copy of BHD without a germline mutation would support a tumor suppressor function for BHD according to the Knudson two-hit hypothesis.
To address the mutation status of the BHD gene in tumors from Birt-Hogg-Dubé patients, we analyzed a panel of 77 renal tumors by direct DNA sequence analysis. Tumor samples, as well as matched normal samples, were obtained from 12 affected members of BHD families after renal surgery at the National Cancer Institute (NCI). The protocol was approved by the institutional review board of NCI, and the patients gave written informed consent.
BHD patients were often found to have bilateral, multifocal tumors and underwent staged bilateral partial nephrectomies, providing tumor samples for this study. Out of 77 tumors, 45 frozen tumor samples were sectioned and microdissected manually to obtain both tumor and histologically normal tissue. DNA was isolated in a solution containing 10 m M Tris-HCl (pH 8.0), 1 m M EDTA, 1% Tween 20, and 0.1 mg/mL proteinase K as described ( 19 ) . For the remaining 32 tumors, tissue was no longer available for microdissection, and DNA was obtained from bulk extraction of tissue under high-salt conditions (Puregene kit, Gentra Systems, Minneapolis, MN).
The entire coding region of BHD (exons 4–14) was sequenced in each tumor sample, following polymerase chain reaction (PCR) amplification. PCR was carried out using genomic primers for the BHD gene as previously described ( 17 ) . A modified primer, SKA3–1, with the sequence TTCCTGCCGGTTTTGAAGGTG, was substituted for primer SKA3 because it produced more reliable amplification. Reactions were carried out in a 25 μL volume using Taq PCR Master Mix (Qiagen, Valencia, CA), 0.4 μ M concentrations of each primer, and a volume of input tumor DNA that was empirically determined to result in sufficient product under standard PCR conditions. Sequencing was carried out using Big Dye Terminator chemistry and run on an ABI Prism 310 Genetic Analyzer (PE/Applied Biosystems, Foster City, CA). Each mutation was verified by repeat PCR and sequencing. To confirm a somatic BHD mutation in a tumor sample independent of the second gene copy, PCR amplicons that contained a somatic BHD mutation were cloned into the TOPO TA cloning vector or the TOPO XL cloning vector (Invitrogen, Carlsbad, CA) and characterized by sequencing as described above.
The known germline mutation from each patient was detected in every tumor. In addition, either a somatic mutation or loss of heterozygosity (LOH) was detected in at least one of the tumors in all 12 BHD patients. In total, 54 of 77 (70%) of their tumors were found to have a somatic mutation or LOH, and, at most, only a single alteration was found in each tumor ( Table 1 ; Fig. 1 ). In contrast, no somatic mutations were found in any matched normal DNA. The somatic mutations that were observed in 41 of 77 (53%) tumors were distributed throughout the BHD coding region, and the majority (30 of 41, 75%) of somatic mutations were frameshift mutations within the open reading frame. Three nonsense mutations were also identified, one in exon 12 and two in exon 14. Interestingly, no missense mutations were detected in this tumor panel.
Tumors exhibiting germline and somatic mutations in Birt-Hogg-Dubé kidney tumors
| Patient ( n ) * | Tumor | Histology † | MD ‡ | Germline mutation § | Somatic mutation § | 1st aa affected |
|---|---|---|---|---|---|---|
| 1 (16) | 2R | Hybrid | No | IVS9+2T>G | c.2065delG ∥ | Ser537 |
| 51R | Hybrid | No | IVS9+2T>G | c.1733delC | His429 | |
| 52R | Hybrid | No | IVS9+2T>G | c.1884C>T (nonsense) | Arg477 | |
| 2L | Hybrid | No | IVS9+2T>G | c.956delG | Gln167 | |
| 5L | Hybrid | No | IVS9+2T>G | c.2039–2043delCAAAG | Lys529 | |
| 6L | Hybrid | No | IVS9+2T>G | IVS10-5delTTC | Predicted splice mutation | |
| 7L | Hybrid | No | IVS9+2T>G | c.1262delG | Thr270 | |
| 8L | Hybrid | No | IVS9+2T>G | c.1508–1509delCA | Met352 | |
| 11L | Hybrid | No | IVS9+2T>G | c.1733insC | His429 | |
| 14L | Hybrid | No | IVS9+2T>G | IVS6-1G>A | Predicted splice mutation | |
| 16L | Chromophobe | No | IVS9+2T>G | LOH germline mutation locus | ||
| 2 (7) | 2 | Hybrid | Yes | IVS9+2T>G | c.1398–1403delGAGAGTinsAA | Glu315 |
| 4 | Hybrid | Yes | IVS9+2T>G | c.998–1005delCCTCATCA; c.1009–1011delCCT | Tyr181 | |
| 6 | Hybrid | Yes | IVS9+2T>G | c.1859insT | Ser469 | |
| 7 | Hybrid | Yes | IVS9+2T>G | LOH germline mutation locus; LOH SNP | ||
| 10 | Oncocytosis¶ | Yes | IVS9+2T>G | LOH germline mutation locus | ||
| 3 (6) | 1 | Hybrid | No | IVS9+2T>G | LOH germline mutation locus | |
| 2 | Hybrid | No | IVS9+2T>G | c.801delC | Gln116 | |
| 5 | Hybrid | No | IVS9+2T>G | LOH germline mutation locus | ||
| 4 (18) | 2L | Hybrid | Yes | c.1733insC | LOH germline mutation locus; LOH SNP | |
| 3L | Hybrid | Yes | c.1733insC | IVS10–5delTTC | Predicted splice mutation | |
| 4L | Hybrid | Yes | c.1733insC | c.1452–1453delTC | Ser333 | |
| 5L | Hybrid | Yes | c.1733insC | c.1387delC | Pro311 | |
| 8L | Hybrid | Yes | c.1733insC | c.841–842delTG | Leu129 | |
| 9L | Hybrid | Yes | c.1733insC | IVS13–4delTTGTTTTGGTTTTC | Predicted splice mutation | |
| 10L | Hybrid | Yes | c.1733insC | LOH germline mutation locus; LOH SNP | ||
| 12L | Hybrid | Yes | c.1733insC | c.1342delA | Glu297 | |
| 15L | Chromophobe | Yes | c.1733insC | c.1000T>C; c.1005–1007delAAC | Leu182Pro; del Asn184 | |
| 16L | Chromophobe | Yes | c.1733insC | c.497delC | His14 | |
| 18L | Chromophobe | Yes | c.1733insC | c.875delC | Ile141 | |
| 1R | Chromophobe | Yes | c.1733insC | LOH germline mutation locus; LOH SNP | ||
| 2R | Chromophobe | Yes | c.1733insC | c.544delC | Pro30 | |
| 3R | Hybrid | Yes | c.1733insC | c.653–654delGG | Ala67 | |
| 6R | Hybrid | Yes | c.1733insC | c.1494–1495insTC | Lys347 | |
| 5 (21) | 3R | Chromophobe | Yes | c.1733insC | c.2043G>T (nonsense) | Glu530 |
| 4R | Chromophobe | Yes | c.1733insC | c.656delC | Ser68 | |
| 5R | Oncocytoma | Yes | c.1733insC | c.733delC | Pro93 | |
| 6R | Oncocytoma | Yes | c.1733insC | c.1579delT | Trp376 | |
| 7R | Hybrid | Yes | c.1733insC | c.1779delC | His442 | |
| 8R | Hybrid | Yes | c.1733insC | c.2082G>T (nonsense) | Glu543 | |
| 9R | Chromophobe | Yes | c.1733insC | c.1742insA | His429 | |
| 11R | Chromophobe | Yes | c.1733insC | c.1591insA | Arg379 | |
| 12R | Hybrid¶ | Yes | c.1733insC | LOH germline mutation locus | ||
| 1L | Oncocytoma | Yes | c.1733insC | LOH germline mutation locus | ||
| 2L | Hybrid | Yes | c.1733insC | c.683delC | Lys78 | |
| 4L | Chromophobe | Yes | c.1733insC | IVS13+1G>A | Predicted splice mutation | |
| 9L | Hybrid¶ | Yes | c.1733insC | c.991insC; c.956–991dup | Ile180 | |
| 6 (1) | 1 | Hybrid | No | c.1733delC | c.1733insC | His429 |
| 7 (1) | 1 | Chromophobe | No | c.1733insC | c.2044–2053delAGGACACACA | Glu530 |
| 8 (1) | 1 | Chromophobe | No | c.1733insC | IVS10-5delTTC | Predicted splice mutation |
| 9 (2) | 1 | Chromophobe | No | c.2034C>T | LOH germline mutation locus | |
| 4 | Chromophobe | No | c.2034C>T | LOH germline mutation locus | ||
| 10 (1) | 1 | Hybrid | No | c.1378-1405dup | LOH germline mutation locus | |
| 11 (2) | 2 | Chromophobe | No | c.1733insC | c.924–926delTTC | Del Phe157 |
| 12 (1) ∥ | 1 | Clear/sarcomatoid | No | c.1733insC | — | — |
| Patient ( n ) * | Tumor | Histology † | MD ‡ | Germline mutation § | Somatic mutation § | 1st aa affected |
|---|---|---|---|---|---|---|
| 1 (16) | 2R | Hybrid | No | IVS9+2T>G | c.2065delG ∥ | Ser537 |
| 51R | Hybrid | No | IVS9+2T>G | c.1733delC | His429 | |
| 52R | Hybrid | No | IVS9+2T>G | c.1884C>T (nonsense) | Arg477 | |
| 2L | Hybrid | No | IVS9+2T>G | c.956delG | Gln167 | |
| 5L | Hybrid | No | IVS9+2T>G | c.2039–2043delCAAAG | Lys529 | |
| 6L | Hybrid | No | IVS9+2T>G | IVS10-5delTTC | Predicted splice mutation | |
| 7L | Hybrid | No | IVS9+2T>G | c.1262delG | Thr270 | |
| 8L | Hybrid | No | IVS9+2T>G | c.1508–1509delCA | Met352 | |
| 11L | Hybrid | No | IVS9+2T>G | c.1733insC | His429 | |
| 14L | Hybrid | No | IVS9+2T>G | IVS6-1G>A | Predicted splice mutation | |
| 16L | Chromophobe | No | IVS9+2T>G | LOH germline mutation locus | ||
| 2 (7) | 2 | Hybrid | Yes | IVS9+2T>G | c.1398–1403delGAGAGTinsAA | Glu315 |
| 4 | Hybrid | Yes | IVS9+2T>G | c.998–1005delCCTCATCA; c.1009–1011delCCT | Tyr181 | |
| 6 | Hybrid | Yes | IVS9+2T>G | c.1859insT | Ser469 | |
| 7 | Hybrid | Yes | IVS9+2T>G | LOH germline mutation locus; LOH SNP | ||
| 10 | Oncocytosis¶ | Yes | IVS9+2T>G | LOH germline mutation locus | ||
| 3 (6) | 1 | Hybrid | No | IVS9+2T>G | LOH germline mutation locus | |
| 2 | Hybrid | No | IVS9+2T>G | c.801delC | Gln116 | |
| 5 | Hybrid | No | IVS9+2T>G | LOH germline mutation locus | ||
| 4 (18) | 2L | Hybrid | Yes | c.1733insC | LOH germline mutation locus; LOH SNP | |
| 3L | Hybrid | Yes | c.1733insC | IVS10–5delTTC | Predicted splice mutation | |
| 4L | Hybrid | Yes | c.1733insC | c.1452–1453delTC | Ser333 | |
| 5L | Hybrid | Yes | c.1733insC | c.1387delC | Pro311 | |
| 8L | Hybrid | Yes | c.1733insC | c.841–842delTG | Leu129 | |
| 9L | Hybrid | Yes | c.1733insC | IVS13–4delTTGTTTTGGTTTTC | Predicted splice mutation | |
| 10L | Hybrid | Yes | c.1733insC | LOH germline mutation locus; LOH SNP | ||
| 12L | Hybrid | Yes | c.1733insC | c.1342delA | Glu297 | |
| 15L | Chromophobe | Yes | c.1733insC | c.1000T>C; c.1005–1007delAAC | Leu182Pro; del Asn184 | |
| 16L | Chromophobe | Yes | c.1733insC | c.497delC | His14 | |
| 18L | Chromophobe | Yes | c.1733insC | c.875delC | Ile141 | |
| 1R | Chromophobe | Yes | c.1733insC | LOH germline mutation locus; LOH SNP | ||
| 2R | Chromophobe | Yes | c.1733insC | c.544delC | Pro30 | |
| 3R | Hybrid | Yes | c.1733insC | c.653–654delGG | Ala67 | |
| 6R | Hybrid | Yes | c.1733insC | c.1494–1495insTC | Lys347 | |
| 5 (21) | 3R | Chromophobe | Yes | c.1733insC | c.2043G>T (nonsense) | Glu530 |
| 4R | Chromophobe | Yes | c.1733insC | c.656delC | Ser68 | |
| 5R | Oncocytoma | Yes | c.1733insC | c.733delC | Pro93 | |
| 6R | Oncocytoma | Yes | c.1733insC | c.1579delT | Trp376 | |
| 7R | Hybrid | Yes | c.1733insC | c.1779delC | His442 | |
| 8R | Hybrid | Yes | c.1733insC | c.2082G>T (nonsense) | Glu543 | |
| 9R | Chromophobe | Yes | c.1733insC | c.1742insA | His429 | |
| 11R | Chromophobe | Yes | c.1733insC | c.1591insA | Arg379 | |
| 12R | Hybrid¶ | Yes | c.1733insC | LOH germline mutation locus | ||
| 1L | Oncocytoma | Yes | c.1733insC | LOH germline mutation locus | ||
| 2L | Hybrid | Yes | c.1733insC | c.683delC | Lys78 | |
| 4L | Chromophobe | Yes | c.1733insC | IVS13+1G>A | Predicted splice mutation | |
| 9L | Hybrid¶ | Yes | c.1733insC | c.991insC; c.956–991dup | Ile180 | |
| 6 (1) | 1 | Hybrid | No | c.1733delC | c.1733insC | His429 |
| 7 (1) | 1 | Chromophobe | No | c.1733insC | c.2044–2053delAGGACACACA | Glu530 |
| 8 (1) | 1 | Chromophobe | No | c.1733insC | IVS10-5delTTC | Predicted splice mutation |
| 9 (2) | 1 | Chromophobe | No | c.2034C>T | LOH germline mutation locus | |
| 4 | Chromophobe | No | c.2034C>T | LOH germline mutation locus | ||
| 10 (1) | 1 | Hybrid | No | c.1378-1405dup | LOH germline mutation locus | |
| 11 (2) | 2 | Chromophobe | No | c.1733insC | c.924–926delTTC | Del Phe157 |
| 12 (1) ∥ | 1 | Clear/sarcomatoid | No | c.1733insC | — | — |
n is the total number of tumors studied for each patient. aa = amino acid; LOH = loss of heterozygosity; SNP = single nucleotide polymorphism.
Histology was determined by analyzing formalin-fixed, paraffin-embedded tissue (or frozen sections in cases with a ¶).
MD denotes whether or not the sample was microdissected.
Mutation position is determined from the first nucleotide of the folliculin transcript, GenBank accession number AF517523 ( 17 , 18 , 23 – 25 ) . Intronic mutations are designated according to the guidelines set forth in Antonarakis ( 26 ) , and “c.” refers to the numbering of a nucleotide location relating to a reference cDNA sequence.
LOH was not found by sequencing in patient 12; however, LOH was detected by microsatellite analysis (data not shown).
Tumors exhibiting germline and somatic mutations in Birt-Hogg-Dubé kidney tumors
| Patient ( n ) * | Tumor | Histology † | MD ‡ | Germline mutation § | Somatic mutation § | 1st aa affected |
|---|---|---|---|---|---|---|
| 1 (16) | 2R | Hybrid | No | IVS9+2T>G | c.2065delG ∥ | Ser537 |
| 51R | Hybrid | No | IVS9+2T>G | c.1733delC | His429 | |
| 52R | Hybrid | No | IVS9+2T>G | c.1884C>T (nonsense) | Arg477 | |
| 2L | Hybrid | No | IVS9+2T>G | c.956delG | Gln167 | |
| 5L | Hybrid | No | IVS9+2T>G | c.2039–2043delCAAAG | Lys529 | |
| 6L | Hybrid | No | IVS9+2T>G | IVS10-5delTTC | Predicted splice mutation | |
| 7L | Hybrid | No | IVS9+2T>G | c.1262delG | Thr270 | |
| 8L | Hybrid | No | IVS9+2T>G | c.1508–1509delCA | Met352 | |
| 11L | Hybrid | No | IVS9+2T>G | c.1733insC | His429 | |
| 14L | Hybrid | No | IVS9+2T>G | IVS6-1G>A | Predicted splice mutation | |
| 16L | Chromophobe | No | IVS9+2T>G | LOH germline mutation locus | ||
| 2 (7) | 2 | Hybrid | Yes | IVS9+2T>G | c.1398–1403delGAGAGTinsAA | Glu315 |
| 4 | Hybrid | Yes | IVS9+2T>G | c.998–1005delCCTCATCA; c.1009–1011delCCT | Tyr181 | |
| 6 | Hybrid | Yes | IVS9+2T>G | c.1859insT | Ser469 | |
| 7 | Hybrid | Yes | IVS9+2T>G | LOH germline mutation locus; LOH SNP | ||
| 10 | Oncocytosis¶ | Yes | IVS9+2T>G | LOH germline mutation locus | ||
| 3 (6) | 1 | Hybrid | No | IVS9+2T>G | LOH germline mutation locus | |
| 2 | Hybrid | No | IVS9+2T>G | c.801delC | Gln116 | |
| 5 | Hybrid | No | IVS9+2T>G | LOH germline mutation locus | ||
| 4 (18) | 2L | Hybrid | Yes | c.1733insC | LOH germline mutation locus; LOH SNP | |
| 3L | Hybrid | Yes | c.1733insC | IVS10–5delTTC | Predicted splice mutation | |
| 4L | Hybrid | Yes | c.1733insC | c.1452–1453delTC | Ser333 | |
| 5L | Hybrid | Yes | c.1733insC | c.1387delC | Pro311 | |
| 8L | Hybrid | Yes | c.1733insC | c.841–842delTG | Leu129 | |
| 9L | Hybrid | Yes | c.1733insC | IVS13–4delTTGTTTTGGTTTTC | Predicted splice mutation | |
| 10L | Hybrid | Yes | c.1733insC | LOH germline mutation locus; LOH SNP | ||
| 12L | Hybrid | Yes | c.1733insC | c.1342delA | Glu297 | |
| 15L | Chromophobe | Yes | c.1733insC | c.1000T>C; c.1005–1007delAAC | Leu182Pro; del Asn184 | |
| 16L | Chromophobe | Yes | c.1733insC | c.497delC | His14 | |
| 18L | Chromophobe | Yes | c.1733insC | c.875delC | Ile141 | |
| 1R | Chromophobe | Yes | c.1733insC | LOH germline mutation locus; LOH SNP | ||
| 2R | Chromophobe | Yes | c.1733insC | c.544delC | Pro30 | |
| 3R | Hybrid | Yes | c.1733insC | c.653–654delGG | Ala67 | |
| 6R | Hybrid | Yes | c.1733insC | c.1494–1495insTC | Lys347 | |
| 5 (21) | 3R | Chromophobe | Yes | c.1733insC | c.2043G>T (nonsense) | Glu530 |
| 4R | Chromophobe | Yes | c.1733insC | c.656delC | Ser68 | |
| 5R | Oncocytoma | Yes | c.1733insC | c.733delC | Pro93 | |
| 6R | Oncocytoma | Yes | c.1733insC | c.1579delT | Trp376 | |
| 7R | Hybrid | Yes | c.1733insC | c.1779delC | His442 | |
| 8R | Hybrid | Yes | c.1733insC | c.2082G>T (nonsense) | Glu543 | |
| 9R | Chromophobe | Yes | c.1733insC | c.1742insA | His429 | |
| 11R | Chromophobe | Yes | c.1733insC | c.1591insA | Arg379 | |
| 12R | Hybrid¶ | Yes | c.1733insC | LOH germline mutation locus | ||
| 1L | Oncocytoma | Yes | c.1733insC | LOH germline mutation locus | ||
| 2L | Hybrid | Yes | c.1733insC | c.683delC | Lys78 | |
| 4L | Chromophobe | Yes | c.1733insC | IVS13+1G>A | Predicted splice mutation | |
| 9L | Hybrid¶ | Yes | c.1733insC | c.991insC; c.956–991dup | Ile180 | |
| 6 (1) | 1 | Hybrid | No | c.1733delC | c.1733insC | His429 |
| 7 (1) | 1 | Chromophobe | No | c.1733insC | c.2044–2053delAGGACACACA | Glu530 |
| 8 (1) | 1 | Chromophobe | No | c.1733insC | IVS10-5delTTC | Predicted splice mutation |
| 9 (2) | 1 | Chromophobe | No | c.2034C>T | LOH germline mutation locus | |
| 4 | Chromophobe | No | c.2034C>T | LOH germline mutation locus | ||
| 10 (1) | 1 | Hybrid | No | c.1378-1405dup | LOH germline mutation locus | |
| 11 (2) | 2 | Chromophobe | No | c.1733insC | c.924–926delTTC | Del Phe157 |
| 12 (1) ∥ | 1 | Clear/sarcomatoid | No | c.1733insC | — | — |
| Patient ( n ) * | Tumor | Histology † | MD ‡ | Germline mutation § | Somatic mutation § | 1st aa affected |
|---|---|---|---|---|---|---|
| 1 (16) | 2R | Hybrid | No | IVS9+2T>G | c.2065delG ∥ | Ser537 |
| 51R | Hybrid | No | IVS9+2T>G | c.1733delC | His429 | |
| 52R | Hybrid | No | IVS9+2T>G | c.1884C>T (nonsense) | Arg477 | |
| 2L | Hybrid | No | IVS9+2T>G | c.956delG | Gln167 | |
| 5L | Hybrid | No | IVS9+2T>G | c.2039–2043delCAAAG | Lys529 | |
| 6L | Hybrid | No | IVS9+2T>G | IVS10-5delTTC | Predicted splice mutation | |
| 7L | Hybrid | No | IVS9+2T>G | c.1262delG | Thr270 | |
| 8L | Hybrid | No | IVS9+2T>G | c.1508–1509delCA | Met352 | |
| 11L | Hybrid | No | IVS9+2T>G | c.1733insC | His429 | |
| 14L | Hybrid | No | IVS9+2T>G | IVS6-1G>A | Predicted splice mutation | |
| 16L | Chromophobe | No | IVS9+2T>G | LOH germline mutation locus | ||
| 2 (7) | 2 | Hybrid | Yes | IVS9+2T>G | c.1398–1403delGAGAGTinsAA | Glu315 |
| 4 | Hybrid | Yes | IVS9+2T>G | c.998–1005delCCTCATCA; c.1009–1011delCCT | Tyr181 | |
| 6 | Hybrid | Yes | IVS9+2T>G | c.1859insT | Ser469 | |
| 7 | Hybrid | Yes | IVS9+2T>G | LOH germline mutation locus; LOH SNP | ||
| 10 | Oncocytosis¶ | Yes | IVS9+2T>G | LOH germline mutation locus | ||
| 3 (6) | 1 | Hybrid | No | IVS9+2T>G | LOH germline mutation locus | |
| 2 | Hybrid | No | IVS9+2T>G | c.801delC | Gln116 | |
| 5 | Hybrid | No | IVS9+2T>G | LOH germline mutation locus | ||
| 4 (18) | 2L | Hybrid | Yes | c.1733insC | LOH germline mutation locus; LOH SNP | |
| 3L | Hybrid | Yes | c.1733insC | IVS10–5delTTC | Predicted splice mutation | |
| 4L | Hybrid | Yes | c.1733insC | c.1452–1453delTC | Ser333 | |
| 5L | Hybrid | Yes | c.1733insC | c.1387delC | Pro311 | |
| 8L | Hybrid | Yes | c.1733insC | c.841–842delTG | Leu129 | |
| 9L | Hybrid | Yes | c.1733insC | IVS13–4delTTGTTTTGGTTTTC | Predicted splice mutation | |
| 10L | Hybrid | Yes | c.1733insC | LOH germline mutation locus; LOH SNP | ||
| 12L | Hybrid | Yes | c.1733insC | c.1342delA | Glu297 | |
| 15L | Chromophobe | Yes | c.1733insC | c.1000T>C; c.1005–1007delAAC | Leu182Pro; del Asn184 | |
| 16L | Chromophobe | Yes | c.1733insC | c.497delC | His14 | |
| 18L | Chromophobe | Yes | c.1733insC | c.875delC | Ile141 | |
| 1R | Chromophobe | Yes | c.1733insC | LOH germline mutation locus; LOH SNP | ||
| 2R | Chromophobe | Yes | c.1733insC | c.544delC | Pro30 | |
| 3R | Hybrid | Yes | c.1733insC | c.653–654delGG | Ala67 | |
| 6R | Hybrid | Yes | c.1733insC | c.1494–1495insTC | Lys347 | |
| 5 (21) | 3R | Chromophobe | Yes | c.1733insC | c.2043G>T (nonsense) | Glu530 |
| 4R | Chromophobe | Yes | c.1733insC | c.656delC | Ser68 | |
| 5R | Oncocytoma | Yes | c.1733insC | c.733delC | Pro93 | |
| 6R | Oncocytoma | Yes | c.1733insC | c.1579delT | Trp376 | |
| 7R | Hybrid | Yes | c.1733insC | c.1779delC | His442 | |
| 8R | Hybrid | Yes | c.1733insC | c.2082G>T (nonsense) | Glu543 | |
| 9R | Chromophobe | Yes | c.1733insC | c.1742insA | His429 | |
| 11R | Chromophobe | Yes | c.1733insC | c.1591insA | Arg379 | |
| 12R | Hybrid¶ | Yes | c.1733insC | LOH germline mutation locus | ||
| 1L | Oncocytoma | Yes | c.1733insC | LOH germline mutation locus | ||
| 2L | Hybrid | Yes | c.1733insC | c.683delC | Lys78 | |
| 4L | Chromophobe | Yes | c.1733insC | IVS13+1G>A | Predicted splice mutation | |
| 9L | Hybrid¶ | Yes | c.1733insC | c.991insC; c.956–991dup | Ile180 | |
| 6 (1) | 1 | Hybrid | No | c.1733delC | c.1733insC | His429 |
| 7 (1) | 1 | Chromophobe | No | c.1733insC | c.2044–2053delAGGACACACA | Glu530 |
| 8 (1) | 1 | Chromophobe | No | c.1733insC | IVS10-5delTTC | Predicted splice mutation |
| 9 (2) | 1 | Chromophobe | No | c.2034C>T | LOH germline mutation locus | |
| 4 | Chromophobe | No | c.2034C>T | LOH germline mutation locus | ||
| 10 (1) | 1 | Hybrid | No | c.1378-1405dup | LOH germline mutation locus | |
| 11 (2) | 2 | Chromophobe | No | c.1733insC | c.924–926delTTC | Del Phe157 |
| 12 (1) ∥ | 1 | Clear/sarcomatoid | No | c.1733insC | — | — |
n is the total number of tumors studied for each patient. aa = amino acid; LOH = loss of heterozygosity; SNP = single nucleotide polymorphism.
Histology was determined by analyzing formalin-fixed, paraffin-embedded tissue (or frozen sections in cases with a ¶).
MD denotes whether or not the sample was microdissected.
Mutation position is determined from the first nucleotide of the folliculin transcript, GenBank accession number AF517523 ( 17 , 18 , 23 – 25 ) . Intronic mutations are designated according to the guidelines set forth in Antonarakis ( 26 ) , and “c.” refers to the numbering of a nucleotide location relating to a reference cDNA sequence.
LOH was not found by sequencing in patient 12; however, LOH was detected by microsatellite analysis (data not shown).
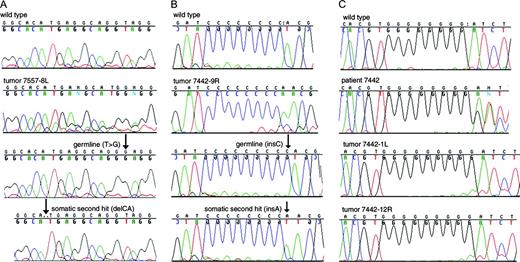
Sequence chromatograms of DNA and clones derived from Birt-Hogg-Dubé tumors. A ) Tumor DNA and clones derived from tumor 8L from patient 1. The top panel depicts the wild type sequence in exon 9. The second panel illustrates DNA directly amplified from tumor 8L. The third panel shows a clone with the IVS9+2T>G germline mutation. The bottom panel depicts a clone containing the somatic 2-bp CA deletion (c.1508–1509delCA) 10 nucleotides upstream, but not the germline mutation. B ) Tumor DNA and clones derived from tumor 9R from patient 5. The top panel shows the wild-type sequence in the C8 tract region of exon 11. The second panel illustrates DNA directly amplified from tumor 9R. The third panel depicts a clone with the germline mutation with a C9 tract (c.1733insC). The bottom panel shows a clone containing the somatic mutation (c.1742insA) but not the germline mutation. C ) LOH demonstrated by sequence chromatograms from direct PCR amplification of BHD tumors. The antisense DNA sequence of a portion of exon 11 is shown. The top panel shows the wild-type sequence from a control DNA sample. The second panel illustrates the normal DNA control from patient 5, showing the overlapping sequences that result from one wild-type copy of BHD and one copy containing the c.1733insC germline mutation. The third panel shows the chromatogram for tumor 1L: only the sequence representing the germline mutation is apparent (note the presence of 9 G peaks). The bottom panel depicts tumor 12R from patient 5; the sequence pattern is similar to tumor 1L.
In addition to the somatic mutations observed in 41 BHD tumors, LOH was identified at the site of the germline mutation or at an observed single nucleotide polymorphism (SNP) in 13 of 77 (17%) tumors ( Table 1 ; Fig. 1 ). In every case of LOH, the germline mutation was retained, and the wild-type sequence was lost. Interestingly, one of patient 2's tumors appeared to have a partial deletion of the gene, because it exhibited LOH at the germline mutation in exon 9 but not at a SNP in intron 12 ( Table 1 ). In all, the frequency of a second hit (LOH or a somatic mutation) in the BHD gene was greater than 50%. Together, these results indicate that renal tumors associated with the BHD syndrome arise from cells in which both copies of the BHD gene are inactivated, supporting the hypothesis that BHD is a tumor suppressor gene.
For BHD to fit the model of a tumor suppressor gene, the second mutation must occur on the wild-type copy of BHD. Although mutation analysis of discrete genomic regions that are amplified by PCR is unable to distinguish between the allele with a germline mutation and the other allele except when the somatic mutation occurs in the same exon as the germline mutation, we observed germline and somatic BHD mutations occurring in the same exon in six of 41 tumors. PCR-amplified products from these exons were subcloned, permitting the identification and sequencing of both BHD alleles. We confirmed that two BHD mutations, one on each allele, had occurred in these renal tumors ( Fig. 1 ). These data, combined with the apparent loss of the wild-type allele in 13 tumors, indicate that the first and second mutations occur on different copies of the BHD gene.
However, not all tumors exhibited LOH or somatic mutations. No LOH or somatic mutations were detected in 23 of 77 tumors (30%). In such cases, gene silencing by hypermethylation or mutations in regulatory regions of the BHD gene may play a role in its inactivation. Alternatively, normal cell contamination of tumor tissue may have interfered with detection of somatic mutations in some cases.
In our study, the frequency of somatic mutations and LOH seen differed slightly by renal tumor histology. Whereas 78% of chromophobe tumors (13 mutations plus five LOH in 23 tumors), 72% (27 mutations and seven LOH in 47 tumors) of oncocytic hybrid tumors, and 50% (three mutations and one LOH in eight tumors) of oncocytomas had second hits, no second hit was detected in either of the two clear-cell tumors. It is unclear whether these differences in mutation and LOH frequencies are due to sampling bias, association of a second hit with histologic type, or differences in the degree of normal cell contamination in these histologic types.
Our data showed that the tumors from a given BHD patient have different second hits. A schematic depiction of kidney lesions from a typical BHD patient with mutations superimposed is shown in Figure 2 . Distinct somatic mutations or LOH were observed in the 15 lesions. This patient also developed several renal tumors with mixed histologic features. Again, LOH and mutation analysis were performed on tissue from the different histologic areas of three such tumors. Within each tumor, the same second hit was observed, regardless of histology. These observations strongly suggest that multiple renal tumors from some BHD patients are independent, clonal events, each arising from a separate and unique second mutation in the BHD gene. However, the three tumors with mixed histologies shared a common somatic mutation in the distinct histologic regions within each tumor. This finding suggests that in some cases, a somatic second hit precedes histologic diversification within a single tumor. The molecular mechanism that drives these events is unknown.
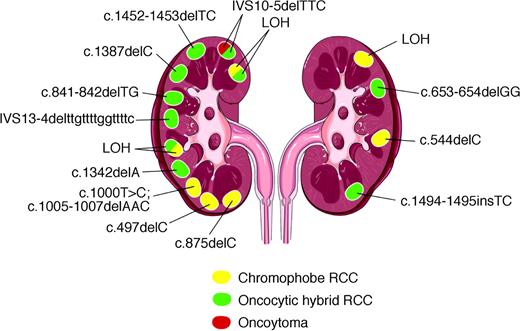
Schematic presentation of tumors and mutations found in the kidneys of patient 4. Of 18 tumors examined, 15 tumors, 11 from the left kidney and four from the right kidney, exhibited a somatic second hit. Tumors with two colors depict those in which areas of different histologic type were microdissected separately from frozen sections; these tumors are categorized from formalin-fixed sections as oncocytic hybrid tumors. Mutations or LOH were observed in all histologic type and in both kidneys.
Interestingly, somatic mutations as the second hit were more common than LOH in BHD tumors. A similar phenomenon is observed in the adenomatous polyposis coli (APC) tumor suppressor gene. The location of the germline APC mutation is thought to influence the type of second hit that occurs ( 20 ). It has been hypothesized that the combination of first and second hits controls and directs an optimal level of β-catenin activity, referred to as the “just right” or “loose fit” model ( 21 , 22 ). To date, there appears to be no relationship between the location of the BHD germline mutation and the nature of the second hit ( Table 1 ). However, tumors with a wider variety of germline mutations should be examined to resolve this question.
In conclusion, this report is the first comprehensive evaluation of a large number of renal tumors from BHD patients with a known germline BHD mutation. Our results document the high frequency and wide spectrum of second mutations, which strongly support a tumor suppressor role for BHD. Inactivation of both copies of BHD occurred in several histologic types of renal tumors, suggesting that BHD may act at an early stage of renal oncogenesis. Further understanding of the mechanism of BHD-induced tumorigenesis awaits functional studies of the folliculin protein.
Present address: Christian P. Pavlovich, Johns Hopkins University, Department of Urology, Baltimore, MD.
This publication has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-C0-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
We thank the BHD patients and their families for their cooperation.
References
Kovacs G, Akhtar M, Beckwith BJ, Bujert P, Cooper CS, Delahunt B, et al. The Heidelberg classification of renal cell tumors.
Storkel S, Eble JN, Adlakha K, Amin M, Blute ML, Bostwick DG, et al. Classification of renal cell carcinoma: Workgroup No. 1. Union Internationale Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC).
Linehan WM, Walther MM, Zbar B. The genetic basis of cancer of the kidney.
Pavlovich CP, Schmidt LS, Phillips JL. The genetic basis of renal cell carcinoma.
Schmidt L, Duh F-M, Chen F, Kishida T, Glenn G, Choyke P, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas.
Schmidt L, Junker K, Kinjerski T, Weirich G, Neumann H, Brauch H, et al. Novel mutations of the MET proto-oncogene in papillary renal carcinomas.
Launonen V, Vierimaa O, Kiuru M, Isola J, Roth S, Pukkala E, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer.
Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer.
Toro JR, Nickerson ML, Wei MH, Warren MB, Glenn GM, Turner ML, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America.
Birt AR, Hogg GR, Dube WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons.
Binet O, Robin J, Vicart M, Ventura G, Beltzer-Garelly E. Fibromes perifolliculaires polypose colique familaile pneumothorax spontanes familiaux.
Toro J, Duray PH, Glenn GM, Darling T, Zbar B, Linehan WM, et al. Birt-Hogg-Dube syndrome: a novel marker of kidney neoplasia.
Zbar B, Alvord G, Glenn G, Turner M, Pavlovich CP, Schmidt LS, et al. Risk of renal and colon neoplasms and spontaneous pneumothorax in the Birt Hogg Dube syndrome.
Pavlovich CP, Hewitt S, Walther MM, Eyler RA, Zbar B, Linehan WM, et al. Renal tumors in the Birt-Hogg-Dube syndrome.
Tickoo SK, Reuter VE, Amin MB, Srigley JR, Epstein JI, Min KW. Renal oncocytosis: a morphologic study of fourteen cases.
Schmidt LS, Warren MB, Nickerson ML, Weirich G, Matrosova V, Toro JR, et al. Birt Hogg Dube syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2.
Nickerson ML, Warren MB, Toro JR, Matrosova V, Glenn G, Turner ML, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome.
Khoo SK, Giraud S, Kahnoski K, Chen J, Motorna O, Nickolov R, et al. Clinical and genetic studies of Birt-Hogg-Dube syndrome.
Zhuang Z, Bertheau P, Emmert-Buck MR, Liotta LA, Gnarra JR, Linehan WM, et al. A microdissection technique for archival DNA analysis of specific cell populations in lesions <1 mm in size.
Lamlum H, Ilyas M, Rowan A, Clark S, Johnson V, Bell J, et al. The type of somatic mutation at APC in familial adenomatous polyposis is determined by the site of the germline mutation: a new facet to Knudson's ‘two-hit’ hypothesis.
Albuquerque C, Breukel C, van der Luijt R, Fidalgo P, Lage P, Slors FJ, et al. The ‘just-right’ signaling model: APC somatic mutations are selected based on a specific level of activation of the beta-catenin signaling cascade.
Crabtree M, Sieber OM, Lipton L, Hodgson SV, Lamlum H, Thomas HJ, et al. Refining the relation between ‘first hits’ and ‘second hits’ at the APC locus: the ‘loose fit’ model and evidence for differences in somatic mutation spectra among patients.
Khoo SK, Kahnoski K, Sugimura J, Petillo D, Chen J, Shockley K, et al. Inactivation of BHD in sporadic renal tumors.
da Silva NF, Gentle D, Hesson LB, Morton DG, Latif F, Maher ER. Analysis of the Birt-Hogg-Dube (BHD) tumour suppressor gene in sporadic renal cell carcinoma and colorectal cancer.
Kahnoski K, Khoo SK, Nassif NT, Chen J, Lobo GP, Segelov E, et al. Alterations of the Birt-Hogg-Dube gene (BHD) in sporadic colorectal tumours.


{kind=link}
{kind=link}