




Abstract
Genetic inactivation of tumor suppressor genes initiates human cancers. However, interaction of accessory cells with the tumor-initiating cell within the microenvironment is often required for tumor progression. This paradigm is relevant to understanding neurofibroma development in neurofibromatosis type I patients. Somatic inactivation of the Nf1 tumor suppressor gene, which encodes neurofibromin, is necessary but not sufficient to initiate neurofibroma development. In contrast, neurofibromas occur with high penetrance in mice in which Nf1 is ablated in Schwann cells in the context of a heterozygous mutant (Nf1+/−) microenvironment. Neurofibromas are highly vascularized, and recent studies suggest that Nf1+/− mice have increased angiogenesis in vivo. However, the function of neurofibromin in human endothelial cells (ECs) and the biochemical mechanism by which neurofibromin regulates neoangiogenesis are not known. Utilizing Nf1+/− mice, primary human ECs and endothelial progenitor cells harvested from NF1 patients, we identified a discrete Ras effector pathway, which alters the proliferation and migration of neurofibromin-deficient ECs in response to neurofibroma-derived growth factors both in vitro and in vivo. Thus, these studies identify a unique biochemical pathway in Nf1+/− ECs as a potential therapeutic target in the neurofibroma microenvironment.
INTRODUCTION
Mutations in the NF1 gene cause neurofibromatosis type I, an autosomal dominant disorder with an incidence of 1 in 3500 (1,2). Neurofibromin, the protein encoded by NF1, functions as a negative regulator of Ras activity (3–5). Neurofibromas are pathognomonic for NF1 (1), and plexiform neurofibromas can give rise to malignant peripheral nerve sheath tumors (MPNSTs) (1,6). Neurofibromas are highly vascularized (7,8), and development of MPNSTs is dependent at least in part on their ability to initiate neoangiogenesis (6,9). Further, recent studies demonstrate that Nf1+/− mice have increased angiogenesis in vivo (10,11) and that disruption of neurofibromin in murine endothelial cells (ECs) recapitulates the cardiovascular defects of the germline Nf1 null phenotype (12). Although these studies demonstrate an essential role of neurofibromin in regulating EC development, the function of neurofibromin in human ECs and the biochemical mechanism by which neurofibromin regulates neoangiogenesis are not known.
Neurofibromas are characterized by excessive extracellular matrix deposition and interactions among different cell types, including Schwann cells, fibroblasts, ECs and mast cells (2,13,14). Recent important insights were gained by analyzing genetically engineered mice that develop plexiform neurofibromas (15). In these studies, conditional deletion of both Nf1 alleles in murine Schwann cells was necessary but not sufficient to generate neurofibromas (15). However, when mice harboring a conditional deletion of Nf1 in Schwann cells were backcrossed onto a Nf1 heterozygous background, the animals developed plexiform neurofibromas (15). These studies demonstrated the essential function of Nf1 heterozygous cells in neurofibroma formation and the importance of understanding the mechanisms of how these cells are recruited to and function within the tumor microenvironment.
The initiation of angiogenesis, which is orchestrated by both the tumor-initiating cell and infiltrating inflammatory cells, is an indispensable step in the neoplastic process (16,17). These observations have led to an interest in identifying drugs that target ECs within the tumors (18–20). Previous studies have shown that Nf1−/− Schwann cells secrete increased concentrations of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF), which are growth factors linked to induction of the ‘angiogenic switch’ required for tumorigenesis (21–24). Further, Nf1+/− mast cells, which are inflammatory cells identified within neurofibromas, secrete increased concentrations of VEGF and bFGF (13,25).
Given the potential importance of ECs and neoangiogenesis to neurofibroma formation, we designed experiments to test whether neurofibromin-deficient ECs have increased angiogenesis in response to growth factors previously identified within neurofibromas and to chemotactic factors secreted by Nf1−/− Schwann cells. In this report, we show that Nf1+/− mice have increased angiogenesis in response to Nf1−/− Schwann cell-derived growth factors in vivo via hyperactivation of the canonical Ras-Raf-Mek-Erk pathway. Further, utilizing primary human vessel wall-derived ECs and endothelial progenitor cells (EPCs) harvested from NF1 patients, we demonstrate that the biochemical mechanism identified for the Nf1+/− murine angiogenesis phenotype is conserved in human cells.
RESULTS
Transduction of human ECs with a shRNA directed against neurofibromin increases activation of the Ras-Erk pathway
Angiogenesis is a complex process involving a sequence of discrete steps (26,27). EC proliferation and migration from pre-existing blood vessels are critical events in establishing a tumor vasculature (26,27). Neurofibromas are highly vascular (7,8), and Nf1−/− Schwann cells secrete both VEGF and bFGF (21–24). However, the function of neurofibromin in regulating human EC function is unknown.
Human microvascular endothelial cells (HMVECs) were transduced with a recombinant retrovirus encoding the selectable marker gene, green fluorescent protein (GFP), and either a shRNA directed against neurofibromin (NF1 shRNA) or a control scrambled oligonucleotide sequence (control shRNA). Following transduction, HMVECs expressing GFP were selected via fluorescence-activated cell sorting (FACS) for experiments (Fig. 1A). HMVECs transduced with the NF1 shRNA expressed reduced levels of neurofibromin by ∼70% post-transduction compared with cells transfected with the control shRNA (Fig. 1B). Further, HMVECs transduced with the NF1 shRNA maintained reduced neurofibromin expression for at least five to six passages (data not shown). Importantly, p120GAP protein levels were not altered in HMVECs transduced with the NF1 or control shRNA (Fig. 1B).
Although neurofibromin functions as a GAP for Ras in some cell lineages (14,28–30), it is unclear whether reduced neurofibromin expression increases Ras activity in primary human ECs. To determine whether neurofibromin functions as a GAP for Ras in primary human ECs, we serum-starved HMVECs transduced with either the NF1 or control shRNA, stimulated the cells with 25 ng/ml VEGF or 50 ng/ml bFGF, and assayed for changes in active Ras-GTP levels. After stimulation, levels of GTP-bound Ras in cellular lysates were determined by precipitating the active GTPase with the GST fusion of the Ras-binding domain of Raf-1 kinase in an effector pull-down assay. HMVECs transduced with the NF1 shRNA had higher basal and VEGF- and bFGF-stimulated Ras-GTP levels when compared with cells transduced with the control shRNA (Fig. 1C). This is consistent with previous studies in Nf1+/− mast cells and Nf1−/− MEFs demonstrating that neurofibromin can function as a basal GAP for Ras (28–30).
We next determined whether shRNA reduction of neurofibromin in ECs alters the activation of either the Erk or PI-3 kinase (PI-3K) effector pathways, which are both linked to EC proliferation and migration (31,32). Serum-starved HMVECs transduced with either the NF1 or control shRNA were stimulated with either 25 ng/ml VEGF or 50 ng/ml bFGF and assayed for changes in Erk and Akt phosphorylation. Activation of Akt is PI-3K-dependent and provides a sensitive measure of PI-3K activity (33,34). After stimulation, cells were lysed and Akt and Erk phosphorylation were measured by western blot. HMVECs transduced with the NF1 shRNA demonstrated increased basal and growth factor-stimulated Erk activation when compared with cells transduced with control shRNA (Fig. 1D and E). Interestingly, we did not detect differences in Akt activation between the NF1 and control shRNA-transduced HMVECs (data not shown), even though PI-3K is hyperactivated in other neurofibromin-deficient cell types (28,35–38). Taken together, these data demonstrate that neurofibromin functions as a negative regulator of the Ras-Erk signaling pathway in primary human ECs.
Increased proliferation and migration of neurofibromin-deficient human ECs is mediated through Erk activation
Proliferation and migration of ECs in response to either VEGF or bFGF is controlled in part by Erk activation (31,32). To determine whether reduced neurofibromin expression enhances EC proliferation and/or migration in response to either VEGF or bFGF, we serum-starved 3×104 HMVECs transduced with the NF1 or control shRNA for 18 h in EBM-2 media without growth factors. Cells were then stimulated with either 25 ng/ml VEGF or 50 ng/ml bFGF for 16 h, and DNA synthesis was measured utilizing tritiated thymidine incorporation assays. HMVECs transduced with the NF1 shRNA had increased basal and growth factor-stimulated proliferation compared with cells transduced with the control shRNA (Fig. 2A).
We next compared the migration of HMVECs transduced with the NF1 or control shRNA in response to either VEGF or bFGF utilizing transwell haptotaxis assays. The 3×104 serum-starved HMVECs were placed in the upper chamber of a collagen-coated transwell. Either EBM-2 culture media without serum and growth factors or 25 ng/ml VEGF or 50 ng/ml bFGF was placed in the lower chamber of the transwell to stimulate migration. After 3 h, migrated HMVECs were counted under a microscope at 20× magnification. In the absence of growth factors, HMVECs transduced with the NF1 shRNA had increased migration compared with HMVECs transduced with the control shRNA (Fig. 2B). Further, HMVECs transduced with the NF1 shRNA had increased migration in response to both VEGF and bFGF compared with cells transduced with the control shRNA (Fig. 2B).
Given that ECs with decreased neurofibromin expression have increased activation of the Ras-Erk pathway, we next tested whether pharmacological inhibition of Erk phosphorylation with the Mek inhibitor, PD98059, would alter the proliferation and/or migration of ECs transduced with the NF1 shRNA in response to either VEGF or bFGF. We compared the migration and proliferation of HMVECs transduced with the NF1 or control shRNA in the presence or absence of 50 µm PD98059 utilizing the migration and proliferation assays described earlier. Both proliferation and migration of HMVECs transduced with the NF1 shRNA was inhibited by PD98059 in response to growth factor stimulation (Fig. 2A and B). Thus, these data demonstrate that genetic reduction of neurofibromin in human ECs enhances signaling through the Ras-Erk pathway to increase EC migration and proliferation in response to VEGF and bFGF.
ECs isolated from NF1 patients have increased proliferation, migration and Erk activation in response to VEGF and bFGF
ECs derived from EPCs can be isolated from adult peripheral blood and expanded ex vivo (39,40). We recently demonstrated that a hierarchy of EPCs exist in adult peripheral blood, which can be discriminated based on their proliferative potential (40). Importantly, we also demonstrated that vessel wall-derived ECs (including HMVECs) contain a complete hierarchy of EPCs similar to EPCs derived from adult peripheral blood (41). Therefore, to test whether the cellular and biochemical phenotype identified in HMVECs transduced with the NF1 shRNA is valid in human NF1 patients, we obtained peripheral blood from NF1 patient and control donors to establish EC cultures for experiments.
To isolate EPCs, we harvested mononuclear cells (MNCs) from NF1 patients and age-/sex-matched healthy donors and cultured the MNCs for outgrowth of EC colonies. EC colonies appeared between 14 and 21 days of cultures as previously described (40), and there was no difference in the number of EC colonies derived from NF1 patients or healthy volunteer donors (data not shown and n=15).
Prior to performing experiments, we verified that the cell progeny derived from EC colonies was not contaminated with hematopoietic cells. This is important because earlier studies have shown that the cell progeny derived from some EC colonies isolated from adult MNCs contain cells, which express the hematopoietic-specific cell surface antigen CD45 (42). After initial EC colony passage, monolayers of spindle shaped cells formed a cobblestone morphology. A representative EC colony is shown in Figure 3A. Immunophenotyping revealed that cells derived from NF1 patients and adult volunteers expressed the EC cell surface-antigens CD31, CD105, CD146, CD144, CD141, vWF and Flk 1 (Fig. 3B) but not the hematopoietic cell-specific surface antigens CD45 or CD14 (Fig. 3B). Thus, these studies confirm that the cell progeny derived from the EC colonies were endothelial in origin and not contaminated with hematopoietic cells. Further, NF1 patient-derived ECs had a ∼70% decrease in neurofibromin expression compared with healthy adult donors (Fig. 4A).
We next determined whether NF1 patient-derived ECs have increased Erk or Akt activation in response to either VEGF or bFGF compared with control ECs. Serum-starved ECs derived from NF1 patient and age-/sex-matched controls were stimulated with either 25 ng/ml VEGF or 50 ng/ml bFGF and assayed for changes in Erk and Akt phosphorylation. NF1 patient-derived ECs demonstrated increased Erk activation compared with control cells (Fig. 4B), but we did not detect differences in Akt activation between the two genotypes (data not shown).
We next determined whether NF1 patient-derived ECs have increased proliferation and/or migration in response to VEGF or bFGF compared with control cells. The 3×104 NF1 patient- or control-derived ECs were serum-starved in EBM-2 media without growth factors. Cells were then stimulated with either 25 ng/ml VEGF or 50 ng/ml bFGF for 16 h, and DNA synthesis was measured utilizing tritiated thymidine incorporation assays. NF1 patient-derived ECs had increased proliferation in response to both VEGF and bFGF compared with cells derived from control donors (Fig. 4C).
We next compared the migration of NF1 patient-derived and control ECs in response to VEGF and bFGF utilizing transwell haptotaxis assays. The 3×104 serum-starved ECs were placed in the upper chamber of a collagen-coated transwell. Either EBM-2 culture media without serum and growth factors or 25 ng/ml VEGF or 50 ng/ml bFGF was placed in the lower chamber of the transwell to stimulate migration. After 3 h, migrated cells were identified and counted as described earlier. In the absence of growth factors NF1 patient-derived ECs demonstrated increased migration compared with control cells. Further, NF1 patient-derived ECs had increased migration in response to both VEGF and bFGF compared with control cells (Fig. 4D).
Given that NF1 patient-derived ECs have increased Erk activation, we next tested whether pharmacological inhibition of Erk phosphorylation with the Mek inhibitor, PD98059, would alter the proliferation and/or migration of NF1 patient-derived ECs in response to either VEGF or bFGF. We compared the migration and proliferation of NF1 patient- and control-derived ECs in the presence or absence of 50 µm PD98059 utilizing migration and proliferation assays described earlier. The proliferation and migration of NF1 patient-derived ECs was inhibited by PD98059 in response to both VEGF and bFGF (Fig. 4C and D). Thus, similar to HMVECs transduced with a NF1 shRNA, NF1 patient-derived ECs have increased migration and proliferation in response to both VEGF and bFGF via Erk hyperactivation.
Nf1+/− mice have an increased angiogenic response to bFGF, VEGF and Nf1−/− Schwann cell conditioned media via hyperactivation of Erk
The matrigel plug assay is a well-established in vivo method to quantitate angiogenesis, which is directly dependent on EC proliferation and migration in response to local administration of different compounds (43). Previous studies have shown that Nf1+/− mice have increased angiogenesis in response to hypoxia and bFGF though the mechanism for this phenotype is unknown (10,11). Given our experimental observations, we hypothesized that local administration of a Mek inhibitor would inhibit the angiogenic response of Nf1+/− mice to VEGF and bFGF in vivo.
To test our hypothesis, we performed matrigel plug assays in WT and Nf1+/− mice in the presence or absence of the Mek inhibitor, PD98059. Implantation of matrigel plugs with pharmacological inhibitors including PD98059 has been previously used to interrogate the function of specific biochemical pathways in initiating angiogenesis (44). Matrigel was mixed with VEGF, bFGF or phosphate-buffered saline (PBS) as a control in the presence or absence of 30 µm of the Mek inhibitor, PD98059, and subcutaneously injected into WT and Nf1+/− mice. Matrigel plugs were harvested 7 days after injection, and the angiogenic response was assessed by visual inspection and quantitation of vessel density by measuring hemoglobin content within the newly formed vessels as previously described (43,44). As shown in Figure 5A and B, Nf1+/− mice have an increased angiogenic response to VEGF and bFGF when compared with WT controls. Enumerating CD31 positive cells by fluorescent antibody staining also confirmed the enhanced angiogenic response of Nf1+/− mice and directly correlated with measurement of hemoglobin content in the matrigel plugs (data not shown). Strikingly, local administration of PD98059 completely inhibited the angiogenic response of both Nf1+/− and WT mice to VEGF and bFGF (Fig. 5A and B).
Finally, given that Nf1−/− Schwann cells, the tumor-initiating cell within neurofibromas, secrete increased concentrations of VEGF and bFGF (21–24), we compared the ability of WT and Nf1−/− Schwann cell conditioned media (SCCM) to induce an angiogenic response in both WT and Nf1+/− mice. Dorsal root ganglia (DRG) were isolated from day 13.5WT and Nf1−/− embryos to establish Schwann cell cultures for collection of CM. Greater than 99% of the WT and Nf1−/− cells were S-100 positive as analyzed by immunohistochemistry (data not shown). WT or Nf1−/− SCCM was mixed with matrigel, injected subcutaneously into WT and Nf1+/− mice, and harvested 7 days later for analysis. As shown in Figure 5C and D, Nf1−/− SCCM markedly increased angiogenesis in Nf1+/− mice when compared with WT controls. However, WT SCCM did not induce a significant angiogenic response in either WT or Nf1+/− mice (Fig. 5C and D). Further, local administration of PD98059 completely inhibited the angiogenic response of Nf1+/− mice to Nf1−/− SCCM (Fig. 6A and B). Taken together, these experiments demonstrate that heterozygous inactivation of Nf1 increases angiogenesis in vivo in response to neurofibroma-derived growth factors and verify that the biochemical mechanisms identified in vitro for the neurofibromin-deficient EC phenotype are operative in vivo.
DISCUSSION
Interaction of stromal supporting cells with the tumor-initiating cell within the microenvironment is often required for tumor progression (15–17). This paradigm is relevant to understanding neurofibroma development in NF1 patients. Genetic studies have demonstrated that Nf1−/− Schwann cells are necessary but not sufficient for neurofibroma formation (15). Specifically, when Zhu et al. used a Krox 20 Cre promoter to induce somatic inactivation of Nf1 in Schwann cells, mice did not develop neurofibromas. However, neurofibromas occur with high penetrance in mice in which Nf1 is ablated in Schwann cells in the context of a heterozygous mutant (Nf1+/−) microenvironment (15). These studies demonstrated the essential function of Nf1 heterozygous cells in neurofibroma formation and the importance of understanding the mechanisms of how these cells are recruited to and function within the tumor microenvironment.
Neurofibromas were initially described as white tumors (1,7,22). However, detailed analysis of neurofibromas revealed that they were highly vascular (7,8) and recent studies suggest that Nf1+/− mice have increased angiogenesis in vivo (10,11). Further, multiple cell types within neurofibromas, including Nf1−/− Schwann cells and Nf1+/− mast cells, secrete growth factors (VEGF and bFGF) (13,21–25), which have been linked to recruitment and proliferation of ECs in other human tumors (16,17,45). Based on these previous observations, studies outlined in this manuscript were designed to determine the biochemical and cellular effects of heterozygous inactivation of Nf1 on human ECs located in the neurofibroma microenvironment in order to identify pre-clinical therapeutic targets.
In this report, we demonstrate that human ECs have increased migration and proliferation in response to bFGF and VEGF via hyperactivation of the Ras-Erk pathway. Interestingly, we did not detect differences in Akt activation between NF1 and control shRNA transduced HMVECs. Ras exists as three primary isoforms (H, N and K-Ras) that preferentially activate different signaling pathways (46–48). The Ras isoforms may be recruited to different locations within cells in response to different stimuli or expressed at varying levels in different cells types resulting in activation of specific pathways. It is possible that neurofibromin may preferentially regulate the different Ras isoforms in ECs. We are actively pursuing these possibilities currently in the laboratory. Further, based on observations made in neurofibromin-deficient human ECs, we also show that Nf1+/− mice have an increased angiogenic response to SCCM, bFGF and VEGF, which is completely inhibited in vivo by pharmacologic inhibition of Erk activation. Therefore, these experiments define a distinct biochemical mechanism for the increased proliferation and migration of human neurofibromin-deficient ECs to growth factors previously identified within neurofibromas.
Although pharmacologic targeting of ECs to inhibit the growth and metastasis of solid tumors is a well-established concept, an emerging paradigm in tumor angiogenesis is the functional importance of pericytes, which cover blood vessels and provide microvascular stability (45,49,50). In support of this concept, several studies have shown that tumor vessels lacking pericytes are more dependent on VEGF for their survival than are vessels invested by pericytes (51) and that pericyte density inversely correlates with the effect of antiangiogenic therapy (52). Additional reports have shown that treating mice genetically engineered to mimic human tumors with both an anti-VEGF drug and Gleevec, which inhibits pericyte recruitment and proliferation by directly interfering with PDGF signaling, was highly efficacious (45).
These previous observations in combination with the data provided in the present study have direct relevance in identifying FDA-approved drugs for treating NF1 patients with neurofibromas for several reasons. First, recent studies have demonstrated that Nf1+/− pericytes have increased proliferation in vivo (11), and it is well established that the proliferation and migration of pericytes in the vascular wall is largely controlled by local secretion of PDGF by accessory cells and ECs in the tumor microenvironment (49,50). Further, PDGF activates the Ras-Erk pathway in pericytes to control both their proliferation and migration (53–55). Second, Nf1−/− SCCM secrete increased concentrations of PDGF, and Nf1+/− fibroblasts, which are another major cell constituent of neurofibromas, are hypersensitive to low concentrations of PDGF via hyperactivation of the Ras-Erk pathway (56,57). Finally, in previous studies, we have demonstrated that Nf1+/− mast cells have increased proliferation and migration via hyperactivation of the c-kit signaling pathway, and recent genetic experiments have suggested that Nf1+/− mast cells may directly contribute to the neurofibroma initiation and progression (13,28). This is an important observation since Gleevec is a potent inhibitor of various mast cell functions and the receptor tyrosine kinase activity of the PDGF receptor, which controls both pericyte and fibroblast proliferation (49,58).
Although the pathogenesis of neurofibroma formation remains complex, the current study in combination with previously published experiments demonstrate that various functions of multiple cell types in the neurofibroma microenvironment are controlled by the activation of few common signaling pathways which can be targeted by currently available FDA approved drugs. Similar to the paradigm established in other tumor models, our studies reveal that the therapeutic use of an anti-VEGF drug or Mek inhibitor in combination with Gleevec, may be particularly efficacious in inhibiting neoangiogenesis and the tumor promoting functions of the different cell lineages resident within the neurofibroma microenvironment.
MATERIALS AND METHODS
Animals
Nf1+/− mice were obtained from Dr Jacks at MIT in a C57BL/6.129 background and backcrossed for 13 generations into the C57BL/6J strain. The Nf1 allele was genotyped by PCR as previously described (13). Experiments were conducted in accordance with a protocol approved by the Indiana University Animal Use and Care Committee.
Peripheral blood samples
Blood samples (80–100 ml) were collected by venopuncture in citrate phosphate dextrose anticoagulant solution from 15 NF1 patients at the Indiana University School of Medicine and adult age-/sex-matched volunteers (6 male, 9 female) ranging in age from 18 to 60 years. The Institutional Review Board at the Indiana University School of Medicine approved all protocols, and informed consent was obtained.
Preparation of MNCs
Anticoagulated blood (80–100 ml) was diluted 1:1 with Hanks buffered salt solution (Invitrogen, Grand Island, NY, USA) then overlayed onto an equivalent volume of Histopaque 1077 (ICN, Costa Mesa, CA, USA). Cells were centrifuged at 740g for 30 min and buffy coat MNCs were collected and washed in PBS (Invitrogen) with 2% fetal bovine serum (FBS; Hyclone, Logan, UT, USA).
Culture of EPCs
EPCs were cultured as previously described (40). Briefly, MNCs were resuspended in EGM-2 medium (Cambrex, Walkersville, MD, USA) supplemented with 10% FBS, 2% penicillin/streptomycin (Invitrogen) and 0.25 µg/ml amphotericin B (Invitrogen) (complete EGM-2) and seeded onto tissue culture plates precoated with rat tail collagen type I (BD Biosciences, Bedford, MA, USA) at 5×107 cells/well at 37°C, 5% CO2 in a humidified incubator. Media was changed daily for 7 days and then every other day until colony formation. Non-adherent cells were discarded with each media change. EPC colonies appeared between 14 and 22 days of culture as previously described (40). Colonies were enumerated using an inverted microscope (Olympus, Lake Success, NY, USA) under 40× magnification.
Immunophenotyping of EPCs
Early passage (2,3) EPCs (5×105) were incubated with primary or isotype control antibody and analyzed by FACs (Becton Dickinson, San Diego, CA, USA) as previously described (40). We used directly conjugated primary murine monoclonal antibodies (all from BD Pharmingen, San Diego, CA, USA, unless indicated) against human CD31-fluorescein isothiocyanate (FITC), human CD14-FITC, human CD45-FITC, human CD146-phycoerythrin (PE), human CD141-FITC (Cymbus Biotechnology, Chandlers Ford, UK), human CD105-APC (Caltag, Burlingame, PA, USA), and human CD144-AlexaFluor 647 (Molecular Probes, Eugene, OR, USA). We used directly conjugated mouse immunoglobulin G1κ (Ig G1κ) for isotype controls. For flk-1, cells were incubated with a biotinylated anti-human KDR (Sigma, St Louis, MI, USA) and streptavidin-APC. For von Willebrand factor (vWF), cells were permeabilized as previously described (40) and incubated with 2 µg/ml anti-human vWF (Dako, Carpenteria, CA, USA) or rabbit Ig isotype control (Dako), then goat anti-rabbit-FITC secondary antibody. Cells were analyzed using the FACS Caliber system and CellQuest software (Becton Dickinson).
HMVEC culture
Human dermal microvascular endothelial cells (HMVECs) were obtained from Cambrex at passage 3. Cells were seeded in tissue culture flasks precoated with type I rat tail collagen in complete EGM-2 supplemented with 10% FBS and cultured as previously described (40).
Murine Schwann cell culture and generation of SCCM
Schwann cells were isolated from WT and Nf1−/− DRG at embryonic day 13.5 as described previously (13). Cultures were stained with an antibody directed against S-100 (Sigma), an acidic, calcium-binding protein present in Schwann cells to verify purity. Cells prepared between passages 1–3 were used for experiments. SCCM was harvested as previously described from cell cultures on passage 2 after 24 h of culture in serum-free DMEM (13).
Generation of siRNA and shRNA constructs
NF1 siRNA oligonucleotides were purchased from Ambion (Austin, TX, USA). NF1 shRNA oligonucleotides (5′-gatccGGACACAATGAGATTAGATTTCTCAAGAGAAAATCTAATCTCATTGTGTCCTTTTTTACGCGTg-3′ sense strand) and (5′-aattcACGCGTAAAAAAGGACACAATGAGATTAGATTTTCTCTTGAGAAATCTAATCTCATTGTGTCCg-3′ antisense strand) were designed based on the siRNA oligonucleotides using the BD Biosciences siRNA Hairpin Oligonucleotide Sequence Designer. NF1 shRNA oligonucleotides were synthesized by Integrated DNA Technologies Inc. (Coralville, IA, USA). All sequences contained the MluI restriction site in the hairpin loop region and BamHI and EcoRI overhang sites for directional cloning. The retroviral vector, RetroQZsGreen, containing the U6 promoter and Zoanthus sp (Zs). GFP driven by the early promoter of cytomegalovirus was obtained from BD Biosciences Clontech (Palo Alto, CA, USA). The oligonucleotides were annealed and ligated into the RetroQZsGreen via the BamHI/EcoRI site. Fusion-Blue competent DH5α Escherichia coli (BD Biosciences) were transformed, and clones with proper insertion of the oligonucleotide into the RetroQ vector were determined by digestion with MluI restriction enzyme. The ZsGreen gene is constitutively expressed, and transfection/transduction efficiency was monitored by flow cytometry. Stable cell lines were selected by sorting for GFP positive cells weekly for 4–5 weeks.
Retroviral packaging
To generate retroviral supernatant containing the NF1 shRNA or control shRNA Ampho 293 packaging cells (BD Biosciences) were plated onto tissue culture plates and transfected the following day with 7.5 µg RetroQZsGreen NF1 shRNA or RetroQZsGreen scrambled shRNA using LipofectAMINE 2000 (Invitrogen) according to manufacturer's instructions. The medium was changed after 4 h and replaced with complete EGM-2 media. Conditioned media was collected every 12 h, clarified by centrifugation at 515g for 5 min, filtered through a 0.45 mm filter and stored at −80°C. The retroviral conditioned media was collected for 3 consecutive days.
Transduction of target cells
HMVECs were seeded into tissue culture plates precoated with retronectin (CH-296, Takara Shuzo, Otsu, Japan) in complete EGM-2 media and transduced with retroviral supernatant diluted 1:1 with complete EGM-2 media in the presence of 8 µg/ml Polybrene (Sigma) for 4–8 h. Media was replaced with complete EGM-2 media overnight. This process was repeated for 3 consecutive days. Transduced cells were subcultured to collagen-coated tissue culture plates and at 1 week post-transduction, cells were sorted by FACS to obtain a population of ZsGreen positive expressing cells containing either the NF1 or scrambled control shRNA.
In vivo matrigel plug assay
Matrigel (BD Biosciences) was thawed at 4°C and kept on ice to maintain a liquid consistency while mixed with 60 units/ml heparin (Sigma) and the following components before injection: bFGF (600 ng/ml, Peprotech, Rocky Hill, NJ, USA), VEGF (300 ng/ml, Peprotech), PD98059 (30 µm, New England Biolabs Inc., Beverly, MA, USA), SCCM or PBS. Matrigel (300 µl) was injected subcutaneously into the murine groin. After 7 days, plugs were harvested, photographed and assayed for total hemoglobin using a hemoglobin assay kit (Pointe Scientific, Lincoln Park, MI, USA) as per manufacturer's protocol.
Thymidine incorporation assays
Cells were deprived of growth factors for 24 h, and then plated in six-well dishes at 3×104 cells/well. HMVECs or EPCs were stimulated with VEGF or bFGF for 16 h in a 37°C, 5% CO2, humidified incubator. Cells were pulse-labeled with 1 µCi/ml of tritiated thymidine (Perkin-Elmer Life Sciences Products, Boston, MA, USA) for 5 h, and β emission was measured (Beckman Coulter Inc., Fullerton, CA, USA) as previously described (40). In some experiments, cells were pre-incubated with 50 µm PD98059 or its vehicle 30 min before addition of cytokines. Assays were performed in triplicate.
Haptotaxis assays
The undersides of transwell cell culture inserts with 8 µm pores (BD Biosciences) were coated with 50 µg/ml type I rat tail collagen and placed into lower chambers containing EBM-2 (Cambrex) alone or in combination with VEGF or bFGF. The 3×104 HMVECs or EPCs were suspended in 100 µl EBM-2 and added to the top of each transwell. In some experiments, cells were incubated with 50 µm PD98059 for 30 min before exposure to cytokines. Cells were incubated for 3 h in a 37°C, 5% CO2 humidified incubator. Non-migratory cells were removed with a cotton swab, and migrated cells attached to the bottom surface of the membrane were fixed with methanol at 4°C and stained with hematoxylin (Fisher Scientific Co., Pittsburgh, PA, USA). The average number of migrated cells per higher-power field was counted with an inverted microscope under 20× magnification. As a control, cell migration on BSA was determined and measured less than 0.001% of the total cell population (data not shown). Assays were performed in triplicate.
Western blotting
Erk activation was determined by depriving cells of serum and growth factors for 16–20 h, followed by stimulation with VEGF or bFGF. Neurofibromin expression was examined in subconfluent HMVECs or EPCs. Cells were lysed in non-ionic lysis buffer [20 mm Tris–HCl (Sigma), 137 mm NaCl (Sigma), 1 mm EGTA (Sigma), 1% Triton X-100 (Sigma), 10% glycerol (Sigma), 1.5 mm MgCl2 (Sigma), and complete protease inhibitors (Amersham Pharmacia Biotech, Piscataway, NJ, USA) as described previously (13). Lysates were normalized for protein content using the bicinchoninic acid assay (Pierce Chemical Co., Rockford, IL, USA). Lysates were boiled for 5 min, subjected to SDS-PAGE and transferred to nitrocellulose. The membranes were blocked with PBS containing 5% blotting grade blocker non-fat dry milk (Bio-Rad, Hercules, CA, USA) for 1 h. Membranes were incubated overnight at 4°C with the following antibodies: anti-phospho-Erk-1/2 (Cell Signaling, Beverly, MA, USA), anti-Erk-1/2 (Cell Signaling, Beverly, MA, USA), anti-phospho-Akt (Cell Signaling), anti-Akt (Cell Signaling), anti-P120 (Cell Signaling), β-actin (Sigma) or anti-neurofibromin (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). Secondary antibodies used were either anti-rabbit or anti-mouse IgG conjugated to HRP (Amersham Pharmacia Biotech). Proteins were visualized by ECL (Amersham Pharmacia Biotech).
Ras activation assay
Transduced HMVECs were deprived of serum and growth factors for 18–24 h and stimulated with 25 ng/ml VEGF or 50 ng/ml bFGF. Ras activation was determined using Ras activation assay kits (Upstate USA Inc., Charlottesville, VA, USA) according to the manufacturer's protocol and as described previously (28).
ACKNOWLEDGEMENTS
We thank Janice Walls for her expert administrative assistance in preparation of the manuscript. Grant support: 1 KO8 CA096579-01 (D.A.I.), P50 NS052606 (D.A.I.), NF043019 Department of Defense (D.A.I.), W81XWH-05-1-0161 Department of Defense (F.C.Y.), Riley Children's Foundation (D.A.I., F.C.Y.), P30 CA82709 (D.A.I., F.C.Y.).
Conflict of Interest statement. The authors do not declare any conflict of interest.
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
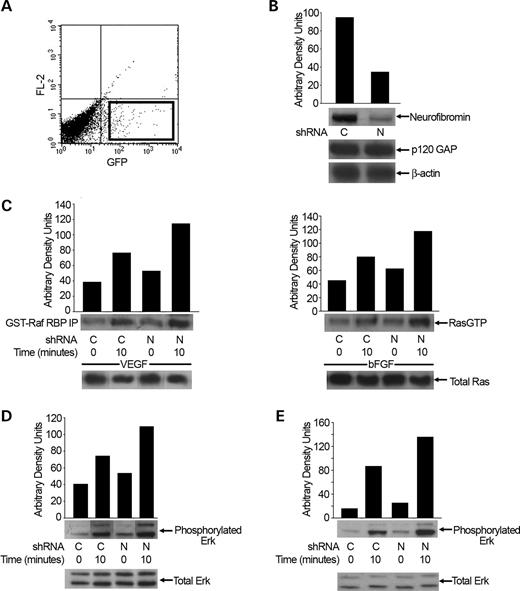
Figure 1. Effect of decreased neurofibromin expression on Ras and Erk activation of human ECs in response to 25 ng/ml VEGF or 50 ng/ml bFGF. (A) HMVECs transduced with RetroQZsGreen NF1 shRNA or control shRNA were selected for GFP expression by FACS. The square box depicts the population of ECs that are GFP positive and thus expressing the shRNA of interest (control or NF1). This population was sorted by FACS and was used for all subsequent experiments. (B) Western blot analysis of neurofibromin and p120GAP expression in HMVECs transduced with NF1 or control shRNA. Data represent one of four independent experiments. C, control shRNA and N, NF1 shRNA. (C) Analysis of Ras activity in HMVECs transduced with NF1 or control shRNA in response to VEGF and bFGF. Immunoblots and quantitative densitometry for RasGTP levels are shown. Western blots for total Ras were used to normalize RasGTP levels and are shown to demonstrate equal loading. Data represent one of four independent experiments. (D and E) Erk activity of HMVECs transduced with control or NF1 shRNA in response to VEGF (D) and bFGF (E). Western blots and quantitative densitometry for Erk phosphorylation are shown. Western blots for total Erk were used to normalize Erk phosphorylation levels and are shown to demonstrate equal loading. Data represent one of four independent experiments.
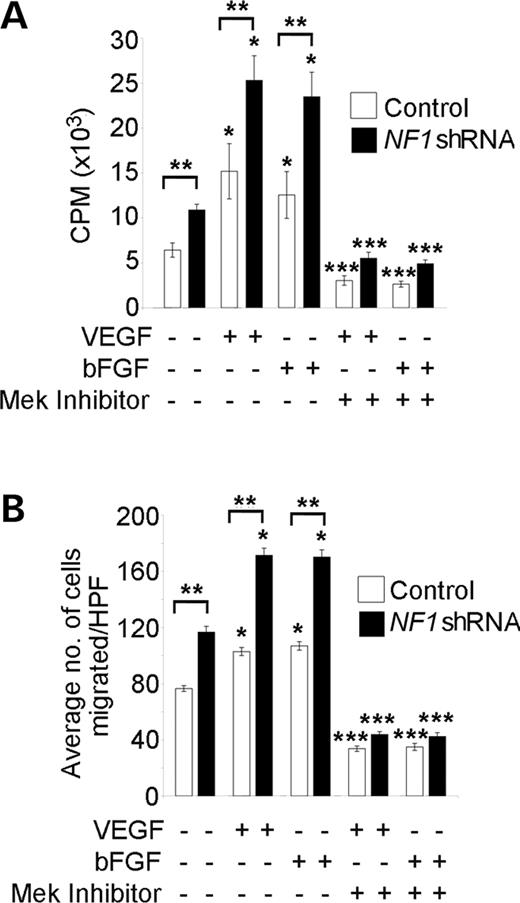
Figure 2. Effect of decreased neurofibromin expression on the proliferation and migration of human ECs in response to 25 ng/ml VEGF or 50 ng/ml bFGF. (A) Proliferation of HMVECs transduced with NF1 shRNA (closed bars) or control shRNA (open bars) in response to VEGF or bFGF in the presence or absence of 50 µm PD98059 (CPM, 3H thymidine counts per minute). *P<0.03 for HMVECs transduced with control or NF1 shRNA stimulated with bFGF or VEGF versus non-stimulated HMVECs; **P<0.02 for HMVECs transduced with NF1 shRNA versus HMVECs transduced with the control shRNA in the absence of growth factor stimulation and in response to bFGF or VEGF; ***P<0.001 for HMVECs transduced with control or NF1 shRNA stimulated with bFGF or VEGF in the presence of PD98059 versus HMVECs transduced with control or NF1 shRNA stimulated with bFGF or VEGF alone, respectively by a Student's paired t-test. (n=4). (B) Haptotaxis of HMVECs transduced with the NF1 or control shRNA in response to bFGF or VEGF in the presence or absence of 50 µm PD98059. Data represent the mean number of migrated cells per ten high power fields±SEM. *P<0.002 for HMVECs transduced with control or NF1 shRNA stimulated with bFGF or VEGF versus non-stimulated HMVECs; **P<0.01 for HMVECs transduced with NF1 shRNA versus HMVECs transduced with control shRNA in the absence of growth factors and in response to either bFGF or VEGF; ***P<0.01 for HMVECs transduced with control or NF1 shRNA stimulated with bFGF or VEGF in the presence of PD98059 versus HMVECs transduced with the control or NF1 shRNA stimulated with bFGF or VEGF alone, respectively, by a Student's paired t-test (n=4).
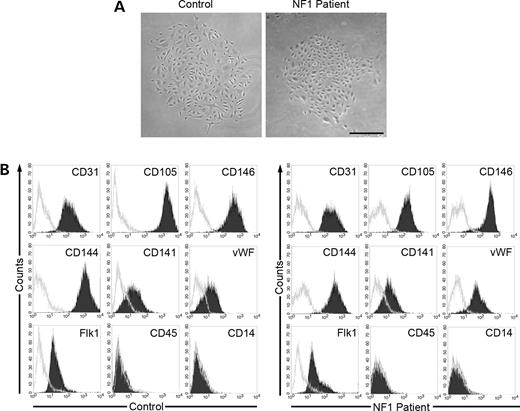
Figure 3. Phenotypic analysis of EC colonies from peripheral blood of adult NF1 patients and controls. (A) Representative photomicrographs of EPC colonies derived from NF1 patient or adult control peripheral blood. Similar colonies were observed from 14 other adult NF1 patients and control donors. Scale bar=100 µm. (B) Immunophenotyping of EC monolayers derived from NF1 patient or adult control EPC colonies by fluorescence cytometry. NF1 patient and adult control EPC-derived ECCs express CD31, CD141, CD105, CD146, CD144, VWF and Flk-1, but do not express CD45 and CD14. Shown are representative data from 15 different NF1 patient and control cell monolayers with similar results. Isotype controls are overlaid in a gray line on each histogram for each surface antigen.
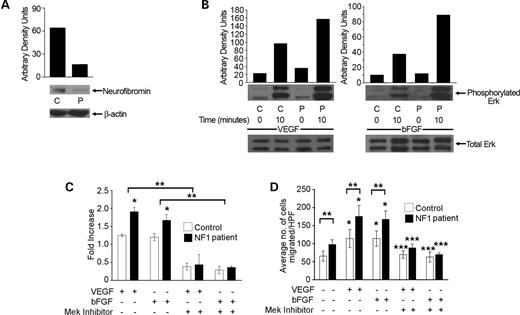
Figure 4. Effect of decreased neurofibromin expression on Erk activation, proliferation and migration of ECs derived from NF1 patients. (A) Western blot analysis of neurofibromin expression in NF1 patient ECs and adult control ECs. Data represent one of four independent experiments. C, adult control ECs; P, NF1 patient ECs. (B) Analysis of Erk activity in NF1 patient or control ECs in response to 25 ng/ml VEGF or 50 ng/ml bFGF. Western blots and quantitative densitometry for Erk phosphorylation are shown. Western blots for total Erk were used to normalize Erk phosphorylation levels and are shown to demonstrate equal loading. Data represent one of four independent experiments. (C) Proliferation of NF1 patient (closed bars) or control ECs (open bars) in response to 25 ng/ml VEGF or 50 ng/ml bFGF in the presence or absence of 50 µm PD98059. There was no difference in proliferation of NF1 patient and control ECs in the absence of growth factors (data not shown). Response to 25 ng/ml VEGF or 50 ng/ml bFGF stimulation in the presence or absence of 50 µm PD98059 is expressed as fold increase over baseline for NF1 patient and control ECs. *P<0.04 for NF1 patient ECs versus adult control ECs in response to bFGF or VEGF; **P<0.006 for NF1 patient or adult control ECs stimulated with bFGF or VEGF in the presence of PD98059 versus NF1 patient or adult control ECs stimulated with bFGF or VEGF alone, respectively, by a Student's paired t-test (n=5). (D) Haptotaxis of NF1 patient ECs and control ECs in response to 25 ng/ml VEGF or 50 ng/ml bFGF in the presence or absence of 50 µm PD98059. Data represent the mean number of migrated cells per 10 high power fields±SEM. *P<0.02 for NF1 patient or adult control ECs stimulated with bFGF or VEGF versus non-stimulated NF1 patient or adult control ECs; **P<0.03 for NF1 patient ECs versus adult control ECs in the absence of growth factors and in response to bFGF or VEGF; ***P<0.04 for NF1 patient or adult control ECs stimulated with bFGF or VEGF in the presence of PD98059 versus NF1 patient or adult control ECs stimulated with bFGF or VEGF alone, respectively, by a Student's paired t-test (n=4).
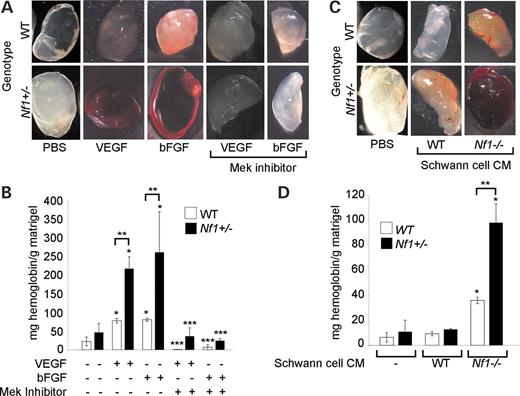
Figure 5. Effect of Nf1 heterozygosity on angiogenesis in vivo. (A) Photomicrographs of matrigel plugs containing 300 ng/ml VEGF, 600 ng/ml bFGF or PBS in the presence or absence of 50 µm of PD98059 harvested from Nf1+/− or WT mice. The bloody appearance of the matrigel plug is a qualitative representation of the in-growth of blood vessels in response to the angiogenic stimuli (VEGF or bFGF). Results represent five independent experiments. (B) Quantification of the angiogenic response of Nf1+/− and WT mice to insertion of matrigel plugs containing VEGF, bFGF or PBS in the presence or absence of PD98059. Results are expressed in milligrams of hemoglobin/gram of matrigel±SEM. *P<0.01 for Nf1+/− or WT with PBS plug versus Nf1+/− or WT with either VEGF or bFGF plugs, respectively; **P<0.02 for WT with either a VEGF or bFGF plug versus Nf1+/− with either a VEGF or bFGF plug, respectively; ***P<0.01 for Nf1+/− or WT with either a VEGF or bFGF plug versus Nf1+/− or WT mice with either VEGF or bFGF in the presence of PD98059, respectively, by a Student's paired t-test (n=5). (C) Photomicrographs of matrigel plugs containing either WT or Nf1−/− SCCM or PBS harvested from Nf1+/− and WT mice. Results are representative of five independent experiments. (D) Quantification of the angiogenic response of Nf1+/− and WT mice to insertion of matrigel plugs containing either WT or Nf1−/− SCCM or PBS. Results are expressed in milligrams of hemoglobin/gram of matrigel±SEM. *P<0.005 for Nf1+/− or WT with PBS plug versus Nf1+/− or WT with Nf1−/− SCCM plugs, respectively; **P<0.02 for WT with Nf1−/− SCCM plugs versus Nf1+/− with Nf1−/− SCCM plugs by a Student's paired t-test (n=5).
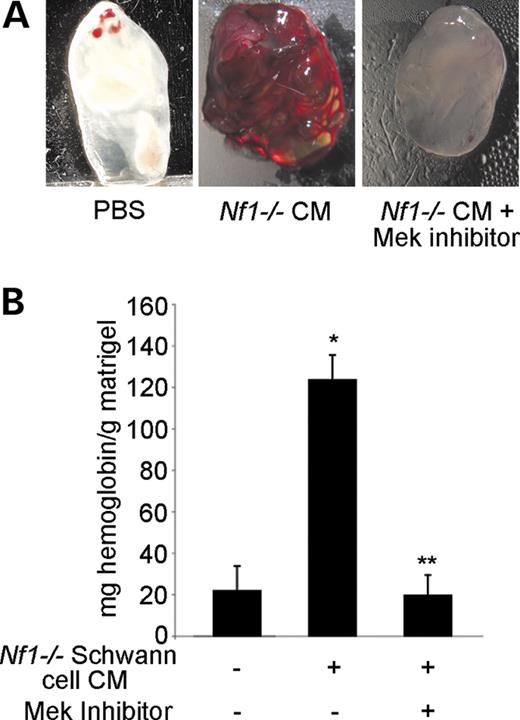
Figure 6. Effect of Mek inhibition on the angiogenic response of Nf1+/− mice to Nf1−/− SCCM. (A) Photomicrographs of matrigel plugs containing either Nf1−/− SCCM or PBS in the presence or absence of 50 µm PD98059 harvested from Nf1+/− mice. The bloody appearance of the matrigel plug is a qualitative representation of the in-growth of blood vessels in response to the angiogenic stimuli (VEGF or bFGF). Results are representative of five independent experiments. (B) Quantification of the angiogenic response of Nf1+/− mice to insertion of matrigel plugs containing either PBS or Nf1−/− SCCM in the presence or absence of 50 µm PD98059. Results are expressed in milligrams of hemoglobin/gram of matrigel±SEM. *P<0.004 for Nf1+/− with a PBS plug versus Nf1+/− with Nf1−/− SCCM plugs (n=5); **P<0.003 for Nf1+/− with Nf1−/− SCCM plugs versus Nf1+/− with Nf1−/− SCCM and PD98059 plugs by a Student's paired t-test (n=5).
References
Friedman, J.M., Gutmann, D.H., MacCollin, M. and Riccardi, V.M. (
Riccardi, V.M. (
Boguski, M.S. and McCormick, F. (
Ballaster, R., Marchuk, D., Boguski, M., Saulino, A., Letcher, R., Wigler, M. and Collins, F. (
Xu, G., O'Connell, P., Viskochil, D., Cawthon, R., Robertson, M., Culver, M., Dunn, D., Stevens, J., Gesteland, R., White, R. et al. (
Angelov, L., Salhia, B., Roncari, L., McMahon, G. and Guha, A. (
Arbiser, J.L., Flynn, E. and Barnhill, R.L. (
Wolkenstein, P., Mitrofanoff, M., Lantieri, L., Zeller, J., Wechsler, J., Boui, M., Revuz, J., Mansat, E. and Stalder, J.F. (
Feldkamp, M.M., Gutmann, D.H. and Guha, A. (
Wu, M., Wallace, M.R. and Muir, D. (
Ozerdem, U. (
Gitler, A.D., Zhu, Y., Ismat, F.A., Lu, M.M., Yamauchi, Y., Parada, L.F. and Epstein, J.A. (
Yang, F.C., Ingram, D.A., Chen, S., Hingtgen, C.M., Ratner, N., Monk, K.R., Clegg, T., White, H., Mead, L., Wenning, M.J. et al. (
Cichowski, K. and Jacks, T. (
Zhu, Y., Ghosh, P., Charnay, P., Burns, D.K. and Parada, L.F. (
Mueller, M.M. and Fusenig, N.E. (
Longo, R., Sarmiento, R., Fanelli, M., Capaccetti, B., Gattuso, D. and Gasparini, G. (
Kerbel, R. and Folkman, J. (
Kim, H.A., Ling, B. and Ratner, N. (
Kawachi, Y., Xu, X., Ichikawa, E., Imakado, S. and Otsuka, F. (
Kurtz, A. and Martuza, R.L. (
Mashour, G.A., Ratner, N., Khan, G.A., Wang, H.L., Martuza, R.L. and Kurtz, A. (
Hirota, S., Nomura, S., Asada, H., Ito, A., Morii, E. and Kitamura, Y. (
Hanahan, D. and Folkman, J. (
Folkman, J. and D'Amore, P.A. (
Ingram, D.A., Hiatt, K., King, A.J., Fisher, L., Shivakumar, R., Derstine, C., Wenning, M.J., Diaz, B., Travers, J.B., Hood, A. et al. (
van der Geer, P., Henkemeyer, M., Jacks, T. and Pawson, T. (
Hiatt, K., Ingram, D.A., Zhang, Y., Bollag, G. and Clapp, D.W. (
Cross, M.J. and Claesson-Welsh, L. (
Larrivee, B. and Karsan, A. (
Datta, K., Bellacosa, A., Chan, T.O. and Tsichlis, P.N. (
Bos, J.L. (
Donovan, S., See, W., Bonifas, J., Stokoe, D. and Shannon, K.M. (
Dasgupta, B., Yi, Y., Chen, D.Y., Weber, J.D. and Gutmann, D.H. (
Johannessen, C.M., Reczek, E.E., James, M.F., Brems, H., Legius, E. and Cichowski, K. (
Hiatt, K., Ingram, D.A., Huddleston, H., Spandau, D.F., Kapur, R. and Clapp, D.W. (
Lin, Y., Weisdorf, D.J., Solovey, A. and Hebbel, R.P. (
Ingram, D.A., Mead, L.E., Tanaka, H., Meade, V., Fenoglio, A., Mortell, K., Pollok, K., Ferkowicz, M.J., Gilley, D. and Yoder, M.C. (
Ingram, D.A., Mead, L.E., Moore, D.B., Woodard, W., Fenoglio, A. and Yoder, M.C. (
Ingram, D.A., Caplice, N.M. and Yoder, M.C. (
Walker, J.M. (Ed.) (
Bhagwat, S.V., Petrovic, N., Okamoto, Y. and Shapiro, L.H. (
Bergers, G., Song, S., Meyer-Morse, N., Bergsland, E. and Hanahan, D. (
Yan, J., Roy, S., Apolloni, A., Lane, A. and Hancock, J.F. (
Walsh, A.B. and Bar-Sagi, D. (
Plowman, S.J. and Hancock, J.F. (
Pietras, K., Sjoblom, T., Rubin, K., Heldin, C.H. and Ostman, A. (
Saharinen, P. and Alitalo, K. (
Benjamin, L.E., Golijanin, D., Itin, A., Pode, D. and Keshet, E. (
Gee, M.S., Procopio, W.N., Makonnen, S., Feldman, M.D., Yeilding, N.M. and Lee, W.M. (
Mourani, P.M., Garl, P.J., Wenzlau, J.M., Carpenter, T.C., Stenmark, K.R. and Weiser-Evans, M.C. (
Zhan, Y., Kim, S., Izumi, Y., Izumiya, Y., Nakao, T., Miyazaki, H. and Iwao, H. (
Huang, J. and Kontos, C.D. (
Cichowski, K., Santiago, S., Jardim, M., Johnson, B.W. and Jacks, T. (
Dang, I. and DeVries, G.H. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}