
Abstract
We report a 38-year-old patient who presented with pulmonary hypertension and right ventricular dysfunction due to pulmonary artery stenoses as a manifestation of Williams syndrome, mimicking chronic thromboembolic pulmonary hypertension. The patient was treated with balloon angioplasty and stent implantation. Short-term follow-up showed a good clinical result with excellent patency of the stents but early restenosis of the segments in which only balloon angioplasty was performed. These stenoses were subsequently also treated successfully by stent implantation. Stent patency was observed 3 years after the first procedure.
Similar content being viewed by others


Pulmonary artery stenosis presenting in adults is rare. Chronic thromboembolic pulmonary hypertension (CTEPH) is by far the most common cause of pulmonary artery stenosis, with an incidence of 0.1–3.8% following a history of (acute) pulmonary embolism [1, 2]. Stenoses in these patients are not caused by an abnormality of the arterial wall itself, but by intraluminal narrowing as a result of the only partially resolved and organized thromboemboli. CTEPH characteristically shows web-like stenoses and/or filling defects, which can be present in smaller as well as larger pulmonary arteries. Nevertheless, distinct disorders such as large vessel vasculitis, peripheral pulmonary stenosis (PPS), pulmonary artery compression, and pulmonary intima sarcoma [3, 4], may mimic the pulmonary vascular occlusions observed in CTEPH.
In general, however, other clinical features will help with the differential diagnosis. Takayasu arteritis (TA) typically presents with aortic coarctation [3]. In most patients with giant cell arteritis [5] and Behçet disease [6] typical clinical features will accompany the pulmonary artery stenoses. Although PPS is a well-known entity in pediatric patients, it is extremely rare in adults [4]. PPS due to an elastin arteriopathy can be associated with a variety of congenital syndromes such as congenital rubella, Williams, Noonan, Alagille, Silver and Ehlers-Danlos [7]. Incidentally, pulmonary artery compression caused by fibrosing mediastinitis [8], pulmonary artery sarcoma [9] or sarcoidosis [10] may also angiographically mimic CTEPH.
Here, we report Williams syndrome as an unusual cause of pulmonary artery stenoses in a middle-aged woman.
Case Report
In 2003, a 38-year-old woman was referred for analysis of suspected CTEPH. She was mildly mentally retarded, which was said to be the consequence of a congenital rubella infection. Two years before admission to our hospital, she presented with acute dyspnea. Based on a high-probability ventilation-perfusion scintigraphy, bilateral multiple pulmonary emboli were diagnosed. Anticoagulant treatment was instituted, whereupon she recovered promptly and, after discharge, she continued working in a sheltered workshop without any physical complaint.
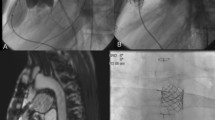
She presented with gradually progressive dyspnea on exertion and a 17 kg weight gain over the preceding few weeks with signs of right ventricular failure. Systemic blood pressure was 130/60 mmHg. Selective pulmonary angiography demonstrated segmental stenoses in both lungs (Fig. 1A, B), Pulmonary artery pressure (PAP) was 114/36 (mean 64) mmHg, mean right atrial pressure 15 mmHg, pulmonary capillary wedge pressure 15 mmHg, cardiac output 3.6 l/min, and the pulmonary vascular resistance 1089 dynes sec cm−5.

A Digital subtraction angiogram of the right pulmonary artery (oblique view) before treatment, demonstrating stenoses at the origin of the right superior lobe trunk, the middle lobe artery, and the lower lobe trunk (arrows). B Digital subtraction angiogram of the left pulmonary artery (oblique view) before treatment, demonstrating occlusion of the left upper lobe and multiple stenoses in the segmental branches of the lower lobe artery (arrows)
Computed tomography (CT) angiography demonstrated a dilated pulmonary trunk and main stem arteries and diffusely narrowed and hypoplastic pulmonary segments (Fig. 2A). A coarctation was demonstrated at the level of the descending and abdominal aorta with a minimal diameter of 8 mm. No other vascular abnormalities were observed. Blood pressure was equal in the two arms and, because of the absence of hemodynamically significant gradient, the coarctation was left untreated.

A Sagittal-oblique thick maximum intensity projection image of the multislice computed tomography angiogram (slice thickness 10 mm) before treatment, showing the stenotic right middle lobe artery (arrowhead). B Repeat computed tomography 6 months after stent placement in the right middle lobe artery in the same projection as in A
In view of the clinical history, CTEPH was considered to be the most likely cause of the pulmonary hypertension. Large vessel vasculitis was considered unlikely. Therefore, a pulmonary endarterectomy (PEA) was offered to the patient. During surgery, instead of chronic thromboembolic lesions, diffuse stenoses of the pulmonary arteries were observed, so PEA could not be performed. A biopsy of the pulmonary artery was obtained. Postoperatively, despite aggressive inotropic support, the patient showed signs of progressive right and left ventricular failure. In view of the absolute necessity to decrease pulmonary vascular resistance, it was decided to perform balloon angioplasty (BA) and stent implantation in the pulmonary arteries. Selective angiography of the left and right pulmonary arteries was performed using a right femoral approach. After placement of a guiding catheter in the pulmonary trunk, balloon angioplasty of four stenoses in segmental arteries was performed, using a 5 mm angioplasty balloon (Cordis OPTA PRO Balloon catheter, Cordis, Johnson & Johnson, Miami Lakes, FL, USA). Immediately thereafter, significant residual stenoses were seen at control angiography in two segmental arteries, the right upper lobe, and right middle lobe. These were subsequently stented with balloon-expandable stents (5 mm/20 mm, Palmaz Genesis, Cordis, Johnson & Johnson, Miami Lakes, FL, USA). In the treated segments no other downstream stenoses were observed. However, one segmental vessel showed a stenosis and an occlusion distal to it. This vessel was left untreated.
Immediately after the procedure, the mean PAP decreased from 61 to 30 mmHg. The clinical course after this procedure was uncomplicated and the patient recovered promptly; she was able to be discharged after 2 weeks. At 3 months follow-up, she was doing well, climbed stairs without complaints, and had resumed work.
The biopsy showed a thickened intima and media of the pulmonary arterial wall with diffuse sclerosis and fibrosis. Genetic analysis was performed and fluorescence in situ hybridization demonstrated a deletion of the long arm of chromosome 7 (7q11.23), pathognomonic for Williams syndrome.
Six months after BA and stent implantation, multislice CT-angiography demonstrated a decrease in the pulmonary trunk from 3.5 to 2.9 cm. Both stents were patent (Fig. 2B). Repeat pulmonary angiography confirmed this observation. However, the pulmonary arterial segments that were treated with BA only showed restenosis. In view of the patient’s good clinical condition, these were left untreated. However, at 24 months follow-up, she again showed signs of right ventricular failure. At this time it was decided to treat the residual stenotic branches of the pulmonary arteries with stent implantation. A similar technique as described before was used. Under general anesthesia, a total of five stents (5 mm/20 mm, Palmaz Genesis, Cordis, Johnson & Johnson, Miami Lakes, FL, USA ) were implanted in four stenotic segmental lesions (one stent in the right lower lobe and four stents in three segments of the left lower lobe). After this procedure, the mean PAP had decreased from 40 to 29 mmHg. The patient recovered well and was discharged 2 days after the procedure. One year later, i.e., 3 years after the initial procedure, control pulmonary angiography showed patent stents in all treated pulmonary segmental branches (Fig. 3A, B). The PAP was 53/18 (mean 32) mmHg. Over the stents in the right and left lower lobes, a pressure gradient of only 3 mmHg was measured.

A, B Digital subtraction angiogram of the right A and left B pulmonary artery (oblique view) 3 years after the initial stenting procedure, showing excellent patency of the stents in the right upper, middle and lower lobe arteries and in the left lower lobe arteries (arrows)
Discussion
In this case report we described a 38-year-old patient who presented with recent-onset pulmonary hypertension due to pulmonary artery stenoses as a manifestation of Williams syndrome mimicking CTEPH. The patient was treated with BA and stenting. Short-term follow-up showed a good clinical result with excellent patency of the stents but early restenosis of the segments in which only BA was performed. All stents were patent after 3 years of follow-up.
In contrast to pediatric patients, in adults with pulmonary artery stenoses BA and stenting are not routinely performed. BA in 12 adult patients with isolated PPS was reported by Kreutzer and coworkers [4]. No specific diagnoses were reported in these patients [4]. Nine of 12 patients were successfully treated, with a follow-up of 52 ± 32 months. Rothman et al.. described 4 cases of adult PPS treated by BA [11]. One patient was free of symptoms for 10 years, whereas in 3 of 4 patients restenosis occurred 4–24 months after BA. They were treated with repeat BA and stent implantation was performed in 2 of the 4. One patient developed stent occlusion, 1 patient died 2 years after stent implantation without other interventions, and 1 patient had an uneventful follow-up for at least 18 months. Furthermore, successful use of BA and stent implantation in incidental cases with pulmonary artery stenoses have been reported [8, 12–15]. BA was also reported to be successful in inoperable CTEPH patients [16]. In Williams syndrome, treatment of pulmonary stenoses with BA and stent implantation has been previously reported in pediatric patients only [17, 18]. Another technique which has been reported in children is the use of cutting-balloon angioplasty, and this seems to yield favorable results at follow-up compared with conventional BA [19, 20]. As cutting balloons have recently become available in larger sizes, up to 8 mm, this technique may become an option for treatment of stenoses in adult-sized arteries.
At 6 months follow-up, we observed restenosis of the BA-treated vessels, whereas all vessels with stent implantation were still patent. This seems consistent with earlier observations in pediatric patients [17, 18]. Relapses after BA therefore appear to be relatively frequent in patients with Williams syndrome. A likely explanation for this is the consistency of the pulmonary artery in these cases. Williams syndrome is a congenital developmental disorder involving the vascular, connective tissue, and central nervous systems that affects 1:20,000 newborns [21]. Deletion of the long arm of chromosome 7 (7q11.23), which encodes for the elastin gene, results in decreased elastin production, affecting the heart, face, and bones. As a consequence, as in our patient, the arterial wall is diffusely thickened, consisting of intimal proliferation and medial dysplasia with hypertrophy, fibrosis, and a mosaic arrangement of smooth muscle cells [17, 18]. This might explain why in these patients stent implantation is superior to BA. Stent patency was observed 3 years after the first procedure. Moreover, the stents did not contribute to the residual pulmonary hypertension observed. However, impaired vascular elastin production without clinical signs of Williams syndrome may affect some (pediatric) patients. This autosomal dominant disorder might be caused by mutations or microdeletions in the elastin gene that are not detected by fluorescence in situ hybridization [22].
In conclusion, we performed a rescue BA and successful multiple stent implantation in a patient who was erroneously diagnosed with CTEPH. Stenting was shown to be superior to BA alone in this patient with Williams syndrome.
References
Fedullo PF, Auger WR, Kerr KM, et al. (2001) Chronic thromboembolic pulmonary hypertension. N Engl J Med 345:1465–1472
Pengo V, Lensing AW, Prins MH, et al. (2004) Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med 350:2257–2264
Kerr KM, Auger WR, Fedullo PF, et al. (1995) Large vessel pulmonary arteritis mimicking chronic thromboembolic disease. Am J Respir Crit Care Med 152:367–373
Kreutzer J, Landzberg MJ, Preminger TJ, et al. (1996) Isolated peripheral pulmonary artery stenoses in the adult. Circulation 93:1417–1423
Brister SJ, Wilson-Yang K, Lobo FV, et al. (2002) Pulmonary thromboendarterectomy in a patient with giant cell arteritis. Ann Thorac Surg 73:1977–1979
Erkan F, Cavdar T (1992) Pulmonary vasculitis in Behçet’s disease. Am Rev Respir Dis 146:232–239
Waller BF, Smith FA, Kerwin DM, et al. (1982) Fetal rubella 27 years later. Chest 81:735–738
Guerrero A, Hoffer EK, Hudson L, et al. (2001) Treatment of pulmonary artery compression due to fibrous mediastinitis with endovascular stent placement. Chest 119:966–968
Routledge T, Jenkins D (2005) Images in clinical medicine. An unusual case of pulmonary embolism. N Engl J Med 352:1578
Battesti JP, Georges R, Basset F, et al. (1987) Chronic corpulmonale in pulmonary sarcoidosis. Thorax 33:76–84
Rothman A, Levy DJ, Sklansky MS, et al. (2003) Balloon angioplasty and stenting of multiple intralobar pulmonary arterial stenoses in adult patients. Catheter Cardiovasc Interv 58:252–260
Fontaine AB, Wood DE, Borsa JJ, et al. (1998) Endovascular treatment of life-threatening peripheral pulmonary artery stenosis. J Vasc Interv Radiol 9:965–967
Mulcahy D, Sigwart U, Somerville J (1991) Successful stenting of a life threatening pulmonary arterial stenosis. Br Heart J 66:463–465
Meckel S, Buitrago-Tellez C, Herrmann R, et al. (2003) Stenting for pulmonary artery stenosis due to a recurrent primary leiomyosarcoma. J Endovasc Ther 10:141–146
Fierro-Renoy C, Velasquez H, Zambrano JP, et al. (2002) Percutaneous stenting of bilateral pulmonary artery stenosis caused by malignant extrinsic compression. Chest 122:1478–1480
Feinstein JA, Goldhaber SZ, Lock JE, et al. (2001) Balloon pulmonary angioplasty for treatment of chronic thromboembolic pulmonary hypertension. Circulation 103:10–13
D’Orsogna L, Sandor GG, Culham JA, et al. (1987) Successful balloon angioplasty of peripheral pulmonary stenosis in Williams syndrome. Am Heart J 114:647–648
Geggel RL, Gauvreau K, Lock JE (2001) Balloon dilation angioplasty of peripheral pulmonary stenosis associated with Williams syndrome. Circulation 103:2165–2170
Bergersen L, Jenkins KJ, Gauvreau K, et al. (2005) Follow-up results of cutting balloon angioplasty used to relieve stenoses in small pulmonary arteries. Cardiol Young 15:605–610
Bergersen L, Perry SB, Lock JE (2003) Effect of cutting balloon angioplasty on resistant pulmonary artery stenosis. Am J Cardiol 15:185–189
Joyce CA, Zorich B, Pike SJ, et al. (1996) Williams-Beuren syndrome: Phenotypic variability and deletions of chromosomes 7, 11, and 22 in a series of 52 patients. J Med Genet 33:986–992
Rodriquez-Revanga L, Badenas C, Carrio A, et al. (2005) Elastin mutation screening in a group of patients affected by vascular abnormalities. Pediatr Cardiol 26:827–831
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Reesink, H.J., Henneman, O.D.F., van Delden, O.M. et al. Pulmonary Arterial Stent Implantation in an Adult with Williams Syndrome. Cardiovasc Intervent Radiol 30, 782–785 (2007). https://doi.org/10.1007/s00270-007-9009-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00270-007-9009-6