





Abstract
The heterogeneity of asthma makes it challenging to unravel the pathophysiologic mechanisms of the disease. Despite the wealth of research identifying diverse phenotypes, many gaps still remain in our knowledge of the disease's complexity. A crucial aspect is the impact of airborne factors over a lifetime, which often results in a complex overlap of phenotypes associated with type 2 (T2), non-T2 and mixed inflammation. Evidence now shows overlaps between the phenotypes associated with T2, non-T2 and mixed T2/non-T2 inflammation. These interconnections could be induced by different determinants such as recurrent infections, environmental factors, T-helper plasticity and comorbidities, collectively resulting in a complex network of distinct pathways generally considered as mutually exclusive. In this scenario, we need to abandon the concept of asthma as a disease characterised by distinct traits grouped into static segregated categories. It is now evident that there are multiple interplays between the various physiologic, cellular and molecular features of asthma, and the overlap of phenotypes cannot be ignored.
Abstract
In asthma patients, host–environment interactions may lead to immune/inflammatory response interplay and phenotype overlap. Asthma should be considered a disorder characterised by a dynamic evolution of phenotypes over a lifetime. https://bit.ly/40kpNWx
Introduction
Asthma is a heterogeneous, complex respiratory disorder characterised by chronic airway inflammation associated with typical respiratory symptoms (dry cough, wheeze, chest tightness and/or dyspnoea), reversible/variable airflow obstruction and airway hyperresponsiveness (AHR). It is, in fact, a collection of multiple obstructive airway disorders with similar symptoms and different clinical presentations. The heterogeneity of asthma is the main feature and clinicians need to confirm the asthma diagnosis and define the correct phenotype based on the clinical history (onset, duration, symptom control, exacerbation, etc.), respiratory functional impairment, biomarkers and the response to treatment.
The first step in asthma phenotyping was taken in the late 1940s when Rackemann [1] distinguished two forms of asthma, namely “extrinsic” (mainly characterised by atopy) and “intrinsic” (not associated with atopy). However, subsequent studies pointed out similarities rather than differences between the two phenotypes [2, 3], which have now largely rendered the classification extrinsic/intrinsic obsolete.
In this context, Robinson et al. [4] characterised two subtypes of T-lymphocytes – T-helper (Th) 1 and Th2 – based on the expression of specific cytokine mRNA patterns, with Th2 cells activated in atopic asthma patients. This work can be considered a turning point as it provided the basis for the definition of Th2 asthma.
The description of the disease has been further complicated by the discovery of a subgroup of severe asthma patients presenting a neutrophilic inflammation of the airways [5, 6], which led to the notion of neutrophilic asthma (NA). Such findings are important for the definition of asthma phenotype based on clinical characteristics and the presence of eosinophils and/or neutrophils in the airways [7, 8].
Since then, respiratory research has described a plethora of phenotypes based on clinical, physiological and pathological parameters, as well as response to treatment and prognostic factors [9].
The initial attempts to phenotype asthma were based on the observation of the clinical and functional characteristics of patients, without considering molecular patterns. Indeed, each phenotype is underlain by multiple interplaying molecular mechanisms (“molecular phenotype”). When a single pathway can account for all the clinical features in a single phenotype, we term it “endotype”, a concept introduced by Anderson [10].
The assessment of induced sputum allowed the definition of different asthma phenotypes according to the inflammation of the airways, namely eosinophilic (sputum eosinophils >2–3%), neutrophilic (sputum neutrophils >61–76%), mixed (both elevated eosinophilia and neutrophilia) and paucigranulocytic asthma (PGA) (both sputum eosinophilia (S-EOS) and neutrophilia under the above cited cut-offs). The key role of induced sputum in phenotyping asthma is testified by the recent work of Hinks et al. [11], which proposed an algorithm to diagnose T2-low asthma in clinical practice that included different approaches, such as sputum cell counts, genetic analysis and volatile organic compounds measurements [12–14].
To obtain an unbiased description of phenotypes, researchers have opted for the statistical approach of cluster analysis. Haldar et al. [15] applied this method to asthmatic populations for the first time and defined clusters of patients sharing distinctive clinical, functional and inflammatory profiles in the so-called Leicester study. Subsequent multicentre studies, the largest being the Severe Asthma Research Program (SARP) and U-BIOPRED, employed different cluster analyses to phenotype different cohorts – the outcomes overlapped to the point that it was possible to make them converge on four groups: 1) early-onset mild allergic asthma (Th2 eosinophilic cluster), 2) early-onset allergic moderate-to-severe remodelled asthma (Th2 mixed granulocytic cluster), 3) late-onset nonallergic eosinophilic asthma (T2 eosinophilic cluster) and 4) late-onset noneosinophilic nonallergic asthma (non-T2 neutrophilic cluster) [16].
The definition of Th2-high and Th2-low asthma derives from the study of Woodruff et al. [17], which found differences in the interleukin (IL)-13-induced gene expression in brushing samples of asthma patients.
A powerful contribution is represented by the discovery of innate lymphoid cells (ILCs) that are stratified into different subsets according to the cytokines secreted and have been shown to be key players, along with Th lymphocytes, in the inflammatory processes occurring in the asthmatic airways [18]. This knowledge prompted a shift in asthma terminology from Th2-high to type (T)2-high asthma.
T2-low asthma is also called non-T2 asthma as several data highlight the multiple non-T2 mechanisms underpinning this molecular phenotype, namely neutrophilic inflammation, Th1/Th17 pathways and neurogenic inflammation.
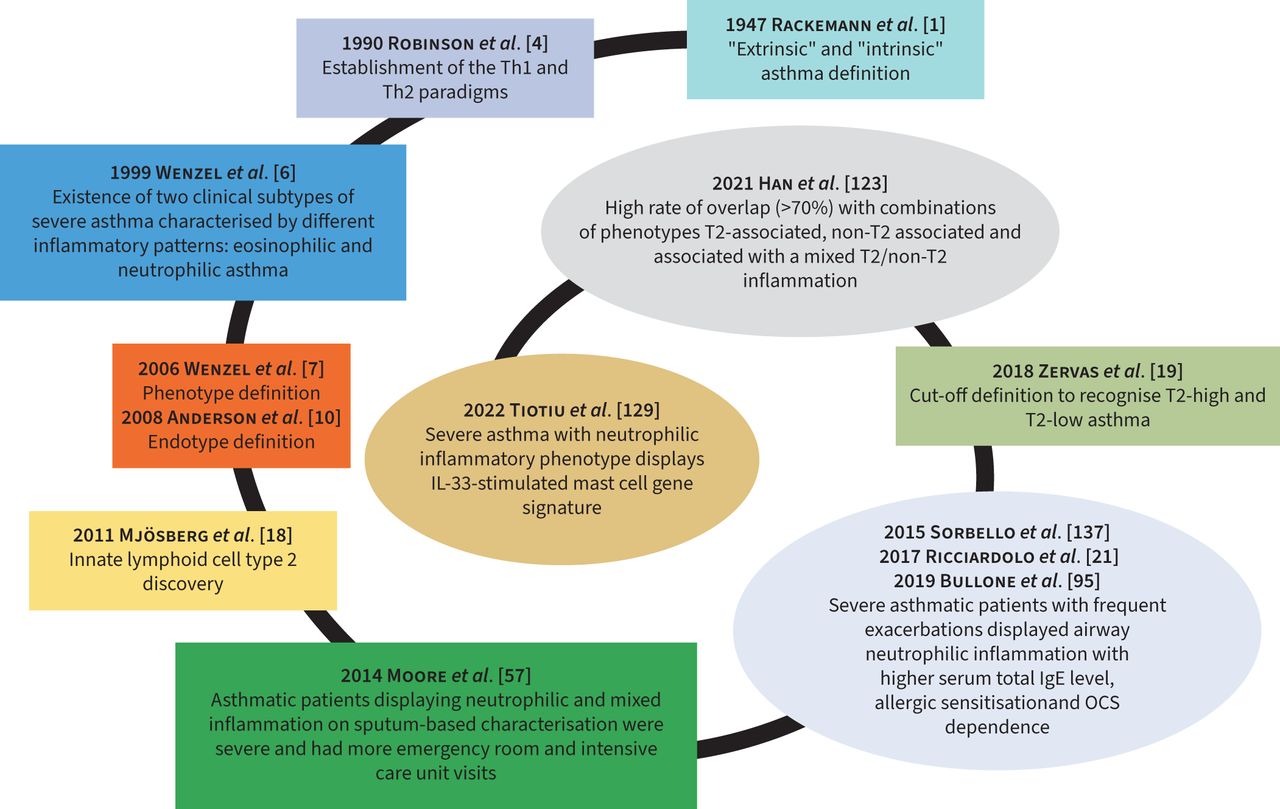
Currently, asthma phenotypes can be included in T2-high and T2-low asthma based on atopic predisposition and at least one of the following biomarkers: elevated serum immunoglobulin E (IgE), high fractional exhaled nitric oxide (FENO) and blood eosinophilia (B-EOS) and/or S-EOS [19]. The main time points in the history of phenotyping asthma are summarised in figure 1.

Snail-shaped spiral graph illustrating the crucial time points (“milestones”) in the asthma history including phenotype overlap. IL: interleukin; OCS: oral corticosteroid; Th: T-helper.
In our opinion, the definition T2-low asthma is more appropriate, since forms of asthma with low S-EOS still have a low-grade inflammation compared to healthy subjects [20]. Consistent with this, increased IL-17 expression in CD4/CD8+ cells in bronchial biopsies from frequent exacerbators is associated with a higher number of both neutrophils and eosinophils [21].
The heterogeneous nature of asthma has led to its designation as an umbrella term rather than a unique disease, encompassing several aspects and traits that can overlap, thus rendering asthma management challenging and demanding accurate phenotyping. This review aims to describe asthma phenotypes and their evolution over time toward a complex network of multiple pathways.
T2-high asthma
Mechanisms
The T2-high phenotypes share an immune–inflammatory response driven by Th2 lymphocytes (adaptive immunity) and group 2 ILCs (ILC2, innate immunity). Hallmarks of T2 inflammation are T2 cytokines such as IL-5, IL-4, IL-13, IL-9, prostaglandin D2 (PGD2) and eosinophils, whose high expression can be detected in the airways (bronchial lumen or wall) and peripheral blood of patients. Indeed, the term eosinophilic asthma is considered a synonym of T2-high asthma, encompasses both allergic and nonallergic phenotypes, and affects about 50% of severe asthma patients [16, 19].
Allergic asthma develops after exposure to different aeroallergens, resulting in the activation of dendritic cells (DCs). DCs in the sub-epithelial layer act as antigen-presenting cells and can recognise and process allergens. After activation, they migrate into the draining lymph nodes where they induce the differentiation of naïve Th cells into Th2 cells, with the cooperation of cytokines (including IL-4) and membrane-expressed molecules (OX40 ligand). Then, Th2 cells move to the lung and direct the Th2 response through the T2 cytokines, promoting class switching of B-cell immunoglobulin to IgE [22–24]. The allergen-specific IgE, cross-linking on the mast cell receptor FcεRI (Fc epsilon receptor I, high-affinity IgE Fc receptor), activates the cells (early phase of allergic response) with the subsequent release from their granules of T2 cytokines, histamine, serotonin, proteases and lipid mediators. Consequently, smooth muscle constriction, increased vascular permeability and recruitment of Th2 cells and eosinophils occur, with increased secretion of cytokines, IgE and activation of eosinophils (late phase of allergic response).
The site of initiation of T2 innate immunity is the epithelium, which in asthma shows altered response mechanisms. After being injured by external stimuli such as pollutants, microbes and allergen-related proteases, the bronchial epithelial cells (BECs) release IL-33, IL-25 and thymic stromal lymphopoietin (TSLP) – alarmins – which exert endowing functions in T2 inflammation. In addition to being overexpressed in the airways of T2-high asthma patients, IL-33 and TSLP regulate Th2 differentiation by interacting with myeloid DCs [25]; a strong correlation of IL-25 expression in T2-high asthma has been found with ATP accumulation and ILC2 expansion in vitro and in human/murine airways [26]. Moreover, the three alarmins can bind to their respective receptors on the ILC2 surface, boosting their activation [24]. In the airways, Th2 lymphocytes, ILC2 and inflammatory cells including eosinophils secrete T2 cytokines, which are involved in recruitment, maturation, survival and activation of eosinophils (eotaxins, IL-5), class switching of B-cell immunoglobulin to IgE, induction of T2 cytokine release (IL-4, IL-13), recruitment of macrophages, and bronchial hyperresponsiveness (IL-9, IL-13) [27]. Furthermore, IL-4 and IL-13 play a pivotal role in the induction of the inducible isoform of nitric oxide synthase in the epithelial layer. This results in nitric oxide production, the concentration of which can be measured in the exhaled breath of asthmatic patients (FENO) and is currently used as biomarker of T2 inflammation [28].
In addition to airway inflammation, structural changes of the airways, broadly referred as to remodelling, accompany T2-high asthma. Among these modifications, goblet cell metaplasia results in a higher production of mucus with the consequent formation of mucus plugs. A recent study by Dunican et al. [29] revealed a more persistent presence of mucus plugs in the airways of severe asthmatic patients with T2 inflammation, eosinophils playing a key role in the formation of the plugs.
An interesting study by Vázquez-Mera et al. [30] reported the relation between T2-high asthma severity and the expression of five serum exosome microRNAs (miRNAs) involved in innate and adaptive immunity regulation. In particular, the expression of miR-21-5p and miR-126-3p, both mediating Th1/Th2 differentiation, were increased in the T2-high-atopic endotype.
In asthma phenotyping, biomarkers represent the logical link from molecular mechanisms to clinical application, as they are required to identify phenotypes, can predict the disease evolution and guide patient-tailored treatment [31]. Currently, the most validated biomarkers of T2-high asthma are B-EOS, S-EOS, FENO and serum total IgE [32–34].
Clinical phenotypes
Early-onset allergic asthma
Frequently, asthma manifestations start during infancy and are triggered by allergens and other environmental stimuli (viral infections, pollutants, oxidants, cigarette smoke) that induce the immune and inflammatory cascade responsible for acute bronchoconstriction. Early-onset asthma (EOA) is typically characterised by atopy, with circulating allergen-specific IgE present, and features the so-called “atopic march”, which is the progression from atopic dermatitis to allergic rhinitis and asthma [35, 36]. In patients with EOA, the cellular processes leading to the T2 signature are orchestrated by Th2 lymphocytes. EOA can present with different degrees of severity and is the most recurrent phenotype in the cluster analyses [16]. It has been hypothesised that the altered immune response initiating and sustaining EOA is underlain by genetic predisposition [36, 37].
EOA can persist throughout life. Studies concerning the determinants of this persistence indicated maternal smoking during pregnancy, upper and lower respiratory tract infections, atopy, rhinovirus infection-induced wheezing, and blood eosinophilia in early life as risk factors for lower lung function, asthma and severe asthma in school-age childhood, adolescence and young adulthood [35, 38–40].
Late-onset nonallergic asthma
Nonallergic asthma begins in adulthood and can be defined as late-onset asthma (LOA) [41]. As well as allergic asthma, LOA is characterised by a prominent T2 immune and inflammatory signature, its distinctive traits being the absence of atopy and consequently IgE signalling and the leading involvement of ILC2. In the lung, ILC2 reside in the mucosal tissue where they participate in immune defence and tissue repair through the release of amphiregulin, which is responsible for epithelial cell proliferation, differentiation and airway remodelling [42, 43]. Two subtypes of ILC2 have been identified: natural ILC2 (nILC2), which lodge in mucosal tissue and take part in its homeostasis, and inflammatory ILC2 (iILC2), which display different surface markers and a higher instability than nILC2 [44]. The activation of ILC2 occurs rapidly due to mediators including alarmins, Toll-like receptor (TLR) ligands (ILC2 express TLR1, 4 and 6), leukotrienes and PGD2 [44]. LOA can manifest with different degrees of severity, the severe form causing lower lung function, higher airflow obstruction and higher eosinophilia despite high-dose inhaled corticosteroid (ICS) treatment [45]. High levels of ILC2 were found in the airways and peripheral blood of severe nonallergic asthma patients, characterised by uncontrolled sputum eosinophilia and steroid resistance, suggesting a mechanism likely involving IL-33 [46].
A role of ILC2 in steroid resistance has been postulated. A recent study in mice and humans described a subset of iILC2 that expressed CD45RO, were elevated in blood and tissues of patients with chronic rhinosinusitis (CRS) and asthma, and correlated with disease severity and steroid resistance. The CD45RO+ILC2 derive from nILC2 after IL-33 and TSLP-driven activation [47]. Moreover, in LOA patients, the exacerbation rate correlates with high B-EOS [48, 49].
Aspirin-exacerbated respiratory disease (AERD)
AERD is found in strict conjunction with respiratory symptoms rapidly triggered after taking aspirin or a nonsteroidal anti-inflammatory inhibitor of cyclo-oxygenase-1 (COX-1). Aspirin reactions, asthma and nasal polyps represent the hallmarks of the syndrome and this association is known as Samter's triad [50, 51]. Either allergic asthma or allergic rhinitis can manifest prior to aspirin hypersensitivity, both in adults and children, but AERD is not considered an allergic disease in that it lacks the production of aspirin-specific IgE [52]. Approximately 8–26% of patients with CRS and nasal polyps have comorbid AERD, while its prevalence is around 7% in all asthmatic patients and twice that in severe asthma patients [53].
In addition to the well-known T2 machinery, AERD is characterised by an overproduction of cysteinyl leukotrienes (CysLts) due to the inhibition of COX-1 and the shift towards the lipo-oxygenase (LOX) pathway, no more inhibited by the prostaglandin E2 (COX-1 product). The chemoattractant effect of CysLts is potentiated, ending up in eosinophil accumulation in the airways. Recently, the involvement of ILC2 in AERD has been postulated. Eastman et al. [54] observed a recruitment of ILC2 from blood to the nasal mucosa, with a concurrent increase in symptoms, during COX-1 inhibitor challenge of AERD patients. White et al. [55] proposed that ILC2 and mast cells could be activated by alarmins derived from epithelial cells after CysLts stimulation leading ILC2 to secrete proinflammatory cytokines which, along with mast cell-derived PGD2, perpetuate and enhance eosinophilic airway inflammation, mucus production, bronchoconstriction and tissue remodelling.
T2-low asthma
Mechanisms
T2-low asthma encompasses all forms of asthma lacking the distinctive traits of T2-high asthma. According to the airway inflammatory profile, we can distinguish neutrophilic (sputum neutrophilia with low eosinophilia), mixed (high sputum neutrophilia and eosinophilia) and paucigranulocytic (low sputum neutrophilia and eosinophilia) asthma. T2-low asthma can differ in severity, with the mixed and neutrophilic phenotypes more often having a higher severity and exacerbation rate [21, 56, 57].
The T1 and T3 immune pathways play main roles in the development of T2-low phenotypes. The T1 response is triggered to counteract viral infections. Viruses can activate TLR on the surface of epithelial cells with the consequent DC release of IL-12, able to induce the differentiation of Th1. This, together with ILC1, CD8+ and natural killer cells, produces interferon gamma (IFN-γ), which is associated with AHR in severe asthma [58].
T3 immunity is mediated by Th17 lymphocytes and ILC3, which secrete cytokines belonging to the IL-17 family (A–F and IL-22) and contributing to steroid resistance, neutrophil recruitment to the airways, mucous cell metaplasia, airway smooth muscle mass hyperplasia and fibroblast proliferation [59]. A study by our group highlighted a higher expression of IL-17F and A in severe asthma compared to mild asthma and controls and in asthma with frequent exacerbations compared to asthma without frequent exacerbations in conjunction with the colocalisation of IL-17F in CD4+ and CD8+ cells in the bronchial mucosa of severe asthma and asthma with frequent exacerbations [21].
Other studies showed the involvement of the multiprotein signalling complex nucleotide-binding oligomerisation domain-like receptor pyrin domain-containing 3 (NLRP3) inflammasome in NA [60]. Inflammasome activation depends either on the interaction between pathogen-associated or damage-associated molecular patterns and TLR or on tumour necrosis factor (TNF) and IL-1β activity. NLRP3 activation results in the upregulated expression of NLRP3, caspase-1, IL-1β and IL-18 [61]. Recent studies reported that NA patients had higher sputum expression of NLRP3, IL-1β and caspase-1. Moreover, the NLRP3 inflammasome–caspase-1–IL-1β pathway in neutrophilic and severe refractory asthma involves a higher expression of NLRP3 and IL-18 receptor genes in neutrophilic and mixed asthma [60, 62, 63].
The investigation carried out by Lachowicz-Scroggins et al. [64] found positive correlations between the levels of extracellular DNA emitted from neutrophil extracellular traps (NETs), neutrophil percentage, myeloperoxidase (neutrophil activation biomarker), NET components, IL-1β and caspase-1 in the sputum of severe asthma patients. In the same study, a primary culture of human BECs released IL-8 and IL-6 after exposure to activated neutrophils.
Serum IL-6 is related to T2-low asthma and obesity [59]. The link with NA is supported by findings that described a subset of asthmatic individuals with higher expression of both IL-6 receptor mRNA and IL-6 protein associated with higher sputum neutrophils and poor lung function [65].
The causative events of airways neutrophilic inflammation comprise bacterial and viral infections, the latter being a trigger for asthma exacerbations. Simpson et al. [66] observed higher exacerbation frequency and a greater incidence of CRS and viral infections in patients with neutrophilic inflammation. A subgroup of these patients also showed airway colonisation of Moraxella catarrhalis, Haemophilus influenzae, Pseudomonas aeruginosa and Staphylococcus aureus.
The latter observation is in line with evidence showing modifications of the microbiota composition of asthmatic airways according to the inflammatory signature, likely influencing the response to antimicrobial and steroid therapies [67, 68]. Indeed, in the sputum of asthmatic patients, the increased presence of the opportunistic pathogens M. catarrhalis and H. influenzae were associated with higher numbers of neutrophils and IL-8 concentrations [67, 69].
As well as for T2-high asthma, the role of miRNAs in the pathogenesis of T2-low asthma has been investigated. A recent study showed an association between the expression of miR-629-3p, miR-223-3p and miR-142-3p and sputum neutrophilia; miR-223-3p and miR-142-3p were related to airway obstruction (forced expiratory volume in 1 s (FEV1)/forced vital capacity ratio) in vivo and had a regulatory effect on inflammation and wound response in vitro [70]. Similarly, Gomez et al. [71] identified miR-223-3p as a component of an miRNA network strongly associated with sputum neutrophilia, reduced FEV1 and quality of life, and increased hospitalisations per year.
When discussing T2-low asthma, we cannot avoid mentioning the effects of corticosteroids, which reduce eosinophil numbers and favour neutrophil survival [72–74]. Therefore, in asthmatic patients exposed to high doses of ICS or/and prolonged oral corticosteroid (OCS) treatment, it can be postulated that corticosteroid intake may induce a noneosinophilic phenotype [74–77]. However, in the UBIOPRED cohort, those with a molecular phenotype encompassing severe eosinophilic asthmatic patients had greater OCS use than those with a neutrophil-associated molecular phenotype [78]. Moreover, several studies on bronchial biopsies showed a decrease or a steady state of neutrophils after corticosteroid treatment [79–82].
PGA
Remodelling plays a key role in T2-low asthma and particularly in PGA, as the distinctive traits of PGA are the lack of inflammation and the occurrence of remodelling events [20, 83]. The better lung function and the reduced frequency of severe refractory asthma of PGA patients, compared to the other asthma phenotypes, is likely due to low/absent inflammation [84], even if anti-inflammatory treatments succeeded poorly in reversing the structural changes associated with PGA [85, 86].
The involvement of neural mediators has been proposed in PGA, as the administration of a nerve growth factor in mice provoked AHR in the absence of airway inflammation [87].
Concerning T2-low biomarkers, to date there are no validated molecules, although studies on blood/airways samples of asthmatic patients are showing encouraging results [12, 21, 57, 88–95].
Clinical phenotypes
Smoking asthma
Both former and current smokers account for 50% of all asthmatic patients. Smoke can impact on the alterations induced by the pathogenic processes related to asthma, leading to a phenotype associated with accelerated decline of lung function, poor asthma control, more frequent exacerbations, diminished quality of life, a higher rate of hospitalisation and a higher risk of developing comorbidities such as cancer [96]. It has also been observed that cigarette smoking in asthma can result in the development of airflow limitation and fixed airflow obstruction, the latter being related to cumulative exposure to >10 pack years [97]. In the airways, cigarette smoke exposure induces oxidative stress with the release of reactive oxygen species (ROS) and reduction of antioxidant enzymes [98, 99], as well as activation of neutrophils, macrophages and CD8+ T-cells, which in turn release ROS, pro-inflammatory mediators and proteases. The resulting damage to lung tissues and impaired epithelial integrity increases the susceptibility of asthmatic patients to microbial infections [100].
Asthma in smokers has been reported in association with T2-low inflammation [101]. A recent study showed that asthmatic smokers with irreversible airflow obstruction (IRAO) had lower lung function, greater airway wall thickness and a higher prevalence of emphysema than nonsmokers with IRAO. Moreover, only asthmatic smokers displayed a correlation between absolute number/% sputum neutrophils (S-NEU) and % emphysema, suggesting a pathogenic role of neutrophils in this phenotype [102].
The steroid resistance associated with smoke and the exclusion of asthmatic smokers from the majority of clinical trials represent significant challenges for the management of this phenotype.
We cannot avoid mentioning that exposure to smoke and emissions from new furniture and open fires (used for cooking in developing countries) have an impact on asthma risk. It has been reported that increases in nitrogen dioxide, sulphur dioxide and ambient ozone are associated with elevation in asthma admission risk for children and augmented hospital admission for asthma in China. Moreover, a recent study showed a correlation between early-life exposure to traffic-related air pollution and a worsening of lung function [103].
Obesity-induced asthma
Obesity is frequently associated with NA, especially with LOA and female gender [11, 104]. A recent analysis confirmed a genetic correlation between obesity and nonatopic LOA, with the involvement of genes encoding for airway remodelling proteins, suggesting them as a therapeutic target [105]. Obese asthma relates to severe forms of asthma, frequently steroid resistant, and to alterations of clinical and functional features, such as AHR, which can be improved by weight loss [106]. Obesity is a pathological condition characterised by systemic inflammation with the contribution of macrophages. Indeed, macrophages are thought to be responsible for adipose tissue inflammation, resulting in the release of TNF-α, leptin and IL-6 in the circulation [107]. IL-6 is a pleiotropic cytokine whose levels have been found elevated in obese patients with NA [108]. A cross-sectional analysis of two asthmatic cohorts revealed higher levels of plasma IL-6 in obese and severe asthmatic patients displaying lower lung function, more frequent exacerbations and poorer asthma control. The observation that among the obese asthmatic patients 62% had low plasma IL-6 levels prompted further comparative assessment of obese/nonobese patients. The authors revealed that high IL-6 levels were associated with severe asthma and metabolic dysfunction both in obese and nonobese patients [109].
In the lung, the effects of IL-6 impact smooth muscle cell proliferation, airway remodelling and Th17 differentiation [107].
Of note, obesity can occur subsequently to early-onset asthma, likely as a consequence of prolonged corticosteroid therapy altering the immune and inflammatory profile and provoking unexpected responses to treatment [110].
Increased respiratory symptoms, lower lung volumes and worse asthma quality of life have been associated with the inverse correlation between an augmented body mass index–asthma–FENO and the arginine/asymmetric dimethyl arginine ratio in LOA. This would indicate the alteration of arginine metabolism occurring in LOA with concomitant obesity as a possible target mechanism for asthma therapy [111].
Asthma in the elderly
Generally, the cut-off for identifying elderly asthma is >65 years [41, 112]. However, asthma persisting since childhood (long-standing asthma, LSA) needs to be distinguished from LOA as LSA is associated with T2-low inflammation, obesity and smoking habit, and not with familiar predisposition and atopy [41, 113]. The higher rate of morbidity and mortality of older asthmatic patients has a multifaceted cause, since comorbidity, under-perception of asthma symptoms, potential cognitive decline and adverse effects from polypharmacy can occur with ageing. Moreover, ageing processes that alter lung structure and physiology impact on those induced by asthma. Age-related modifications include peripheral airway narrowing, alveolar dilation, reduced elastic recoil, greater chest wall rigidity, diminished respiratory muscle strength and decreased lung functionality [41].
The precise pathophysiology of the phenotype is not clear, thus rendering its management very challenging. It can be postulated that the airway neutrophilic infiltration may be driven by either Th1 or Th17 responses, or both. Indeed, older asthmatic patients have elevated CD4+ and CD8+ cells, which produce Th1 cytokines likely inhibiting the Th2 pathway. Available evidence shows that the number of neutrophils in the airways of asthmatic patients increases with both age and age of asthma onset and is related to airflow limitation [114–116]. Further studies are needed to clarify the potential link with corticosteroid resistance in elderly asthma patients.
Another age-related process that could impact on asthma consists of a blunted response of innate and adaptive immunity (immune senescence), leading to a higher susceptibility to pathogen insults and a low-grade systemic inflammation with the release of IL-1β, IL-6 and TNF-α in the circulation, so-called “inflamm-aging” [41, 117].
Post-menopausal asthma – gender and asthma
The influence of gender on asthma incidence and manifestations has been widely reported. In childhood, asthma affects a higher percentage of boys; while in adulthood, the female population presents a higher number of asthmatic patients [118, 119]. In addition to genetics, this discrepancy can be attributed to hormone activity. High oestrogen and progesterone concentrations can modulate the expression and activity of immune cells, inflammatory cytokines and glucocorticoids resulting in the induction of a Th2 response, with eosinophils and FENO increases during the pre-menstrual phase [118–120]. This likely contributes to the increase and worsening in asthma symptoms, exacerbations and hospitalisations during menstrual, peri-menstrual periods and pregnancy.
On the other hand, low levels of female hormones favour a Th1 environment, this being in line with the observed elevation of circulating IL-1, IL-6 and TNF-α post-menopause [120, 121]. Studies on menopausal asthma reported an increased risk of asthma in lean women taking hormone replacement therapy and lower lung function and increased asthma symptoms in post-menopausal women compared to pre-menopausal patients [119]. Further studies are needed to explore the intimate mechanisms of post-menopausal asthma.
Phenotype overlap
Appropriate identification of the asthma phenotype is crucial for disease management and the stratification of patients into distinct subsets represents a useful tool for this purpose. However, the strong link between heterogeneity and asthma, especially severe asthma, can lead to interconnections of multiple molecular pathways and overlaps between different phenotypes, inducing unexpected clinical outcomes. Individuals who develop T2 asthma, especially in infancy, are expected to face several different environmental stimuli during their lifetime, this likely triggering various pathways which can alter the initial T2 predominance (figure 2). The gene–environment–time (GETOMICS) centrality was also highlighted in COPD [122].

Schematic representation of the T2-high and T2-low pathways occurring during the course of life upon exposure to different stimuli. T2-high: after injury, epithelial cells release alarmins (interleukin (IL)-25, IL-33 and thymic stromal lymphopoietin (TSLP)) that activate innate lymphoid cells (ILCs) and dendritic cells (DCs). Upon allergen/antigen uptake, processing and presentation to naïve T-cells, DCs promote the differentiation of naïve T-helper (Th) cells into Th2 lymphocytes. ILC2 and Th2 secrete pro-inflammatory cytokines, exerting key roles in T2 immune response. T2-low: allergens, pollutants, cigarette smoke, viruses and bacteria can damage and stimulate the airway epithelium, which releases TSLP, IL-33, IL-25 and chemokines such as IL-8 acting as neutrophil chemoattractants. Macrophages and DCs elicit the recruitment of neutrophils and the release of pro-inflammatory cytokines by Th17/ILC3 and Th1/ILC1 cells. IFN-γ: interferon-γ; TNF-α: tumour necrosis factor α. Created in BioRender.com.
A very interesting real-life study by Han et al. [123] evaluated phenotype distribution in a population of mild-to-severe asthmatic patients. The authors observed a high rate of overlap (>70%), with combinations of T2-associated, non-T2-associated and associated to a mixed T2/non-T2 inflammation (about half of the patients) phenotypes, the last group showing the worst clinical outcomes. The average age of the patients (younger in the T2 group, intermediate in the mixed one and older in the non-T2 group) may suggest an evolution of the immune-inflammatory status, starting from a canonical T2 signature that gradually changes into a more complex mixed T2/non-T2 profile.
T2 phenotype overlap has been reported for different degree combinations of allergic, eosinophilic and T2-high subtypes, each defined by the cut-off values of allergen-specific IgE (or skin-prick test positivity), B-EOS and FENO, respectively. The allergic phenotype was the most frequent single phenotype, but also the most frequent to be concomitant with the other phenotypes [124, 125]. These outcomes might be due to a T2 phenotype evolution starting with an atopic mechanism persisting after the induction of a ILC2-driven nonallergic machinery.
Concerning T2/non-T2 overlap, an interesting study monitoring the variations of asthma, allergies and comorbidities over time (from 7 to 53 years of age), showed that early-onset persistent asthma and allergies had the greatest risk to develop COPD, while LOA and allergies were associated with a higher risk of multimorbidities, including diabetes, obesity and cardiovascular diseases. The authors hypothesised that asthma and associated comorbidities can have either common underpinning molecular mechanisms or common exposures or common genetic predispositions [126]. These observations are very interesting in relation to the fact that the comorbidities (COPD, gastro-oesophageal reflux disease (GORD), cardiovascular diseases) commonly associated with a T2-low inflammatory signature can develop in patients with an initial T2-high condition.
In this context, a factor worth mentioning is microbial infections that can recur during a lifetime and skew the immune/inflammatory response. It is well known that both Gram-positive and Gram-negative bacteria release enterotoxins that can act as superantigens. S. aureus is a very common human pathogen that has been linked to atopic conditions such as atopic dermatitis, rhinitis, asthma and CRS with nasal polyps [127]. An investigation of the mechanisms guiding the atopic march revealed that, in mice, an epicutaneous sensitisation to ovalbumin and S. aureus enterotoxin B boosted the Th2 inflammation through a Th17–IL-17 pathway. This led to a mixed inflammatory infiltrate in the airways with increases in eosinophils and neutrophils [128].
Recently, Tiotiu et al. [129] performed transcriptomic analyses on asthmatic sputum samples and identified nine gene signatures derived from mast cells exposed to different stimuli. Severe asthma was associated with the enrichment of all nine signatures, while the stratification of patients based on granulocyte counts revealed an association between the IL-33-stimulated mast cells and NA. The genes upregulated in this signature were related to NF-κB and TNF-α signalling, neutrophil degranulation, and responses to bacteria. Interestingly, the bronchoalveolar lavage (BAL) of severe asthma patients presented a high level of both IL-13 and neutrophils, but not eosinophils. This mixed condition was in conjunction with colonisation of the pathogens M. catarrhalis, Haemophilus sp. and Streptococcus sp. [25].
T2 asthma is commonly associated with CRS with nasal polyps, the latter being associated with nonatopic disease [130]. The pathophysiology of nasal polyps is characterised by the colonisation of several micro-organisms, S. aureus being the most frequent [131]. A reduction has been reported of short palate, lung and nasal epithelium clone 1 (SPLUNC1), a protein expressed in the epithelium and sub-mucosa of the nasopharynx and involved in the innate immune defence [132]. SPLUNC1 reduction has also been observed in 3D primary cultures of BECs after exposure to IL-13 [133, 134] and could be associated with the higher susceptibility to infections in patients with asthma and CRS with nasal polyps.
A cluster study performed on the SARP cohort described a cluster (cluster 5) composed mostly of women with LOA, less atopy and long duration of asthma (longest compared to the other four groups). Cluster 5 had the most severe airflow limitation, received high-dose ICS and frequently used systemic corticosteroids, had the highest prevalence of sinus disease, and the highest number of sputum neutrophils [135]. Similarly, Amenlik et al. [45] described a subset of patients displaying severe LOA characterised by nasal polyposis, high values of FENO, persistent airway eosinophilia and blood neutrophilia. In addition, increased sputum eosinophil and blood neutrophil counts were independently associated with disease severity, irrespective of age, sex or asthma duration. The authors theorised that blood neutrophilia might be a characteristic of LOA – it would derive from the event that initiated the disease such as viruses, atypical bacteria or fungi.
Considering these findings, we can postulate that in patients with predisposition or susceptibility, progressive stimuli, occurring over time, can modify the immune and inflammatory profile by triggering or enhancing cellular and molecular mechanisms, and lead to a complex overlap of phenotypes and endotypes. Several investigations have provided evidence of interplay between pathways previously considered to be mutually exclusive (figure 3).

{kind=link}
{kind=link}
{kind=link}
Illustration of phenotype overlap. During the lifetime of asthmatic patients who develop a T2 immune and inflammatory response, diverse environmental stimuli can induce modifications of the pre-existing inflammatory profile ending up in mixed molecular pathways and phenotype overlap. The arrow highlights the dynamic change from the T2-high to the mixed phenotype. FENO: exhaled nitric oxide fraction; IL: interleukin; ILC: innate lymphoid cell; PGD2: prostaglandin D2; α-SMA: α-smooth muscle actin; TGF-β: transforming growth factor-β; Th: T-helper.
Cosmi et al. [136] detected Th17/Th2 clones in the peripheral blood of asthmatic patients and showed that these could originate from Th17 cells able to acquire Th2 features. Along the same lines, Sorbello et al. [137] reported a high expression of IL-17F/A in severe allergic asthma associated with bronchial neutrophilia and number of exacerbations. In addition, the same group showed increased expression of IL-17F+ cells in bronchial mucosa of (allergic/nonallergic) severe asthma and asthma with frequent exacerbations, suggesting a role for IL-17 as a potential biomarker to identify the frequent exacerbation asthma phenotype [13]. Later, we revealed higher levels of serum total IgE, prevalence of perennial allergen sensitisation and OCS dependence in high-NA patients compared to intermediate-NA ones, in conjunction with a higher rate of exacerbations and IL-17F+ cells in bronchial mucosa. Interestingly, the rise in concentration of serum total IgE along with bronchial neutrophil counts suggests a link between T2 immunity and neutrophilic mechanisms [95]. Likewise, a previous study by Moore et al. [57] divided a cohort of asthmatic patients into four clusters distinguished by the progressive severity of asthma. Atopy was present across the groups, including the clusters with a predominance of patients displaying neutrophilic and mixed airway inflammation.
A cross-sectional study suggested the concomitant existence of Th2 and Th17 pathways in a subset of asthmatic patients displaying airway eosinophilia and a high genetic expression of Th17 [138]. Researchers found dual Th2/Th17 T-cells in blood and BAL of stable allergic and severe asthma patients presenting interactions between T3 and T2 pathways [139, 140]. In this pathway network, a role for ILC2 has been shown with the Notch ligand induced conversion of cultured nILC2 into iILC2 producing IL-13 and IL-17 [141].
A very interesting study demonstrated that obesity could affect a consolidated T2 inflammatory status (atopic dermatitis in mice and allergic asthma in obese patients) altering the immune response through the induction of a Th17 signature which becomes additional to the pre-existing Th2. The concomitant pathways can induce resistance to therapy [142].
Of note, overlap was reported also by Hastie et al. [143], who gauged variations of proteins involved in inflammation and immunity in a population of asthmatic patients stratified by their sputum counts. It emerged that increases in T2 proteins were associated with sputum neutrophilia (≥40%). Moreover, the data of this study indicated an overlap between Th2 and Th1 signatures, as suggested by the connected elevations of macrophage inflammatory protein-3α/chemokine ligand 20 and TNF-α in patients with high S-NEU. In line with this, an analysis of the bronchial mucosa of elderly asthmatic patients displayed a higher expression of neutrophils and vascular remodelling factors in concomitance with a higher number of eosinophils [144].
Finally, we should also consider a study performed on humans and a murine model showing the involvement of growth factor interferon regulatory factor 5 in the development of a Th1/Th17 immune response associated with severe asthma and steroid resistance [145].
Conclusions
Over a lifetime, multiple airborne stimuli activate a variety of molecular pathways in asthma (particularly in T2 asthma) and, in consequence, can rearrange the dominance of the molecular pattern (molecular phenotype) in a dynamic and timely way, provoking the development of a different and complex phenotype (phenotype overlap) characterised by poor clinical control, steroid insensitivity and worsening severity.
Clinicians should, therefore, be more cautious in defining the asthma phenotype based on the current biomarkers used in clinical practice and, in addition, should also search for different phenotype overlaps, especially in steroid-insensitive and difficult-to-treat asthma patients. We need to rethink the classical view on asthma phenotype as a rigid and simple disease with variants. Instead, we should now think of asthma as a mutant, flexible, multifaceted, heterogeneous and complex disorder whose pattern rearranges from time to time in response to the host–environment interaction.
Acknowledgements
The authors wish to thank Rosemary Allpress (Veruno, Italy) for editing and proofreading this manuscript.
Footnotes
Provenance: Submitted article, peer reviewed.
Author contributions: F.L.M. Ricciardolo conceptualised the review and V. Carriero performed literature search and wrote the first draft. F. Bertolini provided the figures of the manuscript. All authors were involved in the reviewing, editing, and approval of the final version of the manuscript.
Conflict of interest: F.L.M. Ricciardolo reports grants from Chiesi, grants and personal fees from AstraZeneca, GSK and Sanofi, and personal fees from Novartis outside the submitted work. G. Guida reports personal fees from AstraZeneca, outside the submitted work. F. Bertolini and A. Di Stefano have no conflicts of interest. V. Carriero received a grant from Sanofi.
- Received October 19, 2022.
- Accepted March 23, 2023.
- Copyright ©The authors 2023
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References










