





Abstract
The world of rare interstitial lung diseases (ILDs) is diverse and complex. Diagnosis and therapy usually pose challenges. This review describes a selection of rare and ultrarare ILDs including pulmonary alveolar proteinosis, pulmonary alveolar microlithiasis and pleuroparenchymal fibroelastosis. In addition, monogenic ILDs or ILDs in congenital syndromes and various multiple cystic lung diseases will be discussed. All these conditions are part of the scope of the European Reference Network on rare respiratory diseases (ERN-LUNG). Epidemiology, pathogenesis, diagnostics and treatment of each disease are presented.
Abstract
Many rare and ultrarare ILDs are underdiagnosed. Increasing insight into pathobiology, genetics and disease behaviour have led to better treatment options. Early consultation with or referral to expert centres (ERN-LUNG) is advised. https://bit.ly/3vwYrib
Introduction: an overview of interstitial lung diseases (ILDs)
ILDs comprise over 200 different entities of known and unknown causes characterised by a large variation in clinical manifestations, radiological and pathological patterns, and outcome [1, 2]. ILDs are divided into different categories (figure S1) [3].
A selection of rare or ultrarare ILDs with a higher probability of being faced in clinical practice will be discussed in this review. Although these diseases are called “interstitial lung diseases”, some of them mainly affect the alveolar space, such as in pulmonary alveolar proteinosis (PAP) and pulmonary alveolar microlithiasis (PAM), or the pleura and subpleural lung parenchyma, as in pleuroparenchymal fibroelastosis (PPFE) [4]. We also describe some ultrarare ILDs, including various congenital syndromes (e.g. Hermansky–Pudlak syndrome, HPS) and various multiple cystic lung diseases.
PAP
PAP, first described in 1958 [5], has an incidence of 0.5–1.5 cases per 1 million [6]. Surfactant accumulates in the alveoli due to a defective clearance by alveolar macrophages resulting in different degrees of respiratory insufficiency [7]. Three forms are known (table 1) [8, 9].
Autoimmune PAP
Epidemiology and clinical presentation
Autoimmune PAP is caused by neutralising autoantibodies against granulocyte–macrophage colony-stimulating factor (GM-CSF) [11] that prevent GM-CSF from binding to its receptor, thereby reducing macrophage stimulation and impairing surfactant clearance [12]. Most patients are between 30 and 50 years old when diagnosed [8]. Patients are predominantly male (2:1) and have a smoking history in 80% of cases [8].
The disease usually becomes symptomatic with exertional dyspnoea. In one third of cases, however, patients are asymptomatic [13]. Other common symptoms include cough, fatigue and weight loss [8, 13], and occasionally chest pain or haemoptysis, which is usually a sign of complications [9].
Diagnosis
As clinical examination is generally normal [9], the diagnosis is usually made on the basis of thoracic imaging and bronchoalveolar lavage (BAL) (figure 1).

a) and b) Chest computed tomography scan and X-ray (same date) of a 37-year-old patient with a diagnosis of pulmonary alveolar proteinosis. a) “Crazy paving” pattern with geographical ground-glass opacities and thickened interlobar septa. b) Bi-pulmonary consolidations with punctum maximum in the left centre field without pleural effusion. c) and d) Bronchoalveolar lavage (BAL) from the same 37-year-old patient. c) Typical BAL with milky appearance. d) BAL cytology with foamy macrophages, cellular detritus and periodic acid Schiff-positive extracellular corpuscles.
In chest computed tomography (CT) a “crazy paving” pattern is often present, which consists of reticulation superimposed on ground-glass opacities with a geographic distribution. Healthy and diseased areas are located directly next to each other [14].
Body plethysmography reveals a restrictive ventilatory disorder. Typically, the transfer factor is decreased [13]. Hypoxaemia can be detected especially during exercise and in 30% of patients also at rest [8].
BAL fluid has a milky appearance. BAL cytology may show an increased proportion of lymphocytes [15, 8] and an increased total cell count [16]. The diagnosis is confirmed by the detection of periodic acid Schiff (PAS)-positive extracellular corpuscles and foamy macrophages [15].
In the vast majority of cases, the combination of typical radiology with the results of BAL are sufficient for diagnosis. Occasionally, a transbronchial biopsy might be required, while surgical lung biopsy is mostly unnecessary [5].
A titre of anti-GM-CSF autoantibodies >19 µg·mL−1 has a sensitivity and specificity of almost 100% [17] for the diagnosis of autoimmune PAP, while a titre <10 µg·mL−1 is normal [5, 18].
Differential diagnoses should consider diseases that may be associated with crazy paving patterns on CT, including acute idiopathic pulmonary fibrosis (IPF) exacerbations, acute interstitial pneumonias, acute respiratory distress syndrome, pulmonary oedema, drug-induced pneumonitis and Pneumocystis jirovecii pneumonia [10].
Treatment
The gold standard of treatment is whole lung lavage (WLL), which removes the proteinaceous material from the alveoli, thus restoring oxygenation [19]. The prerequisites are a general anaesthesia and intubation via a double-lumen endotracheal tube. One lung is treated at a time with up to 40 L of warmed saline solution, while the patient is ventilated on the contralateral side. The liquid is applied until it becomes clear. During the procedure, regular bronchoscopic position checks of the tube and physiotherapeutic measures are required [19]. However, depending on the centre, the exact practice (e.g. choice of the first lung to be lavaged, position and volume instilled per lung) shows a great variation [20]. The result of WLL is a significant clinical and radiological improvement [21]. A small case series showed different surfactant components accumulating in the airways after WLL, e.g. increased surfactant protein (SP) A until the second hour with a rapid decrease afterwards [22]. If necessary, the procedure can be repeated contralaterally [9].
Recent pilot studies have investigated the efficacy and tolerability of inhaled and subcutaneous GM-CSF substitution in autoimmune PAP [9, 23]. This was recently confirmed by two large randomised studies [23, 24]. The IMPALA-study was a double-blind, placebo-controlled study with nebulised molgramostim, a recombinant GM-CSF which met the primary end-point, change in Aa-DO2 (alveolar–arterial oxygen gradient) from baseline to 24 weeks. The secondary end-point quality of life also showed significant improvement with continuous, daily molgramostim administration. In addition, molgramostim showed a good safety profile [23]. In the Pulmonary Alveolar Proteinosis GM-CSF Inhalation Efficacy (PAGE) trial, also double-blind and placebo-controlled, sargramostim (recombinant human GM-CSF) treatment was associated with a significant positive change in alveolar–arterial oxygen difference PaO2 (partial pressure of oxygen) [24].
The use of immunosuppressants such as corticosteroids is not recommended [25]. Plasmapheresis can decrease circulating anti-GM-CSF antibodies. However, only very few case reports exist in which the procedure was successful after a previously failed WLL [26], while in other patients this procedure was not successful [9].
In cases of failed WLL, rituximab has been studied in a few small trials [9]. It showed effectiveness in terms of arterial blood oxygenation, CT scans and pulmonary functional parameters. BAL showed lower levels of anti-GM-CSF antibodies, but this could not be confirmed in serum [27, 28]. Another real-life setting retrospective study showed contrasting results [9]. After 6 months, 0/13 patients showed improvement following treatment with rituximab [29]. Rituximab may be considered as an off-label therapy on a case-by-case basis in severe, refractory cases pending availability of inhaled recombinant GM-CSF; however, prospective clinical trial data are not yet available [5].
A case of recurrence after lung transplantation has been documented; nevertheless, lung transplantation can be cautiously considered [9]. With the idea of targeting lipid homeostasis, statin therapy could be taken into consideration; however, with effectiveness in only two patients the evidence is low [30]. In exceedingly rare cases of PAP with progressive fibrosis, antifibrotic therapy might be considered, analogous to the INBUILD-trial [31].
Secondary PAP
Haematological disorders account for the largest proportion (75%) of secondary PAP [32], especially myelodysplastic syndromes and acute myeloid leukaemia [33]. Rarely, PAP occurs in immunodeficiency (e.g. severe combined immunodeficiency, agammaglobulinemia and after organ transplantation) [25]. Treatment of the underlying disease is essential as a cure can be achieved by haematologic therapy, chemotherapy and bone marrow transplantation alone, especially in acute leukaemia [5, 9]. The effect of WLL does not last long in secondary PAP [9].
Inhalation noxious agents include organic and inorganic dusts such as quartz, metal or chemical substances [33]. Relevant dust exposure is also present in autoimmune PAP in up to 50% of cases [8]. Therefore, avoidance is of utmost importance.
Genetic PAP
In genetic PAP, children are most affected and the diseases are associated with a poor prognosis [7, 9]. Mutations predominantly occur in genes involved in the production of surfactant (table 1). Genetic PAP shares features with autoimmune PAP, but differs with respect to radiology and histology.
Mutations with involvement of surfactant production include SFTPB, SFTPC, ABCA3 and NKX2-1. Typical findings are diffuse ground-glass opacities and lung cysts [5]. Mutations can also affect macrophages and the degradation of surfactant [5]. The diseases behave very similarly to autoimmune PAP. There are no anti-GM-CSF antibodies, but high concentrations of GM-CSF [5]. Corticosteroids are the preferred drug and can be combined with other immunomodulators. In advanced cases, lung transplantation must be considered.
Genetic defects of the GM-CSF receptor mainly affect children and result in diseases similar to autoimmune PAP. High concentrations of GM-CSF are detectable, but no antibodies against GM-CSF. WLL can be effective while GM-CSF therapy shows no effects [34].
Mutations in SLC7A7 result in lysinuric protein intolerance, an autosomal-recessive inherited disease. Frequent manifestations are renal and pancreatic insufficiency as well as different pulmonary manifestations like PAP. Management includes WLL and nebulised GM-CSF therapy [34].
Prognosis
With regard to all forms of PAP, Bonella et al. [8] reported an improvement or stabilisation within 12 months with no WLL required in 43 out of 70 patients. In smokers, more WLLs are needed to achieve remission [8]. Secondary PAP is associated with higher mortality, particularly in the case of underlying haematologic diseases [32].
PAM
The first description of PAM dates back to 1918 [35]. The disease is associated with the accumulation of hydroxyapatite microliths within alveolar spaces. With about 1000 cases worldwide, PAM is among the ultrarare lung diseases [36]. It is caused by mutations in the sodium phosphate co-transporter gene SLC34A2, which represents a possible future target for therapies [36].
Epidemiology and clinical presentation
A review of the 1022 cases reported worldwide showed that PAM is present in all continents with a majority in Asia (56.3%) and in Europe (27.8%), and a slight predominance in men. Patients between the ages of 20 and 30 are most commonly affected. Inheritance plays a role in 32–61% of the reported cases, which is autosomal recessive [37].
A mutation in the SLC34A2 gene, described in 2016, results in a defective sodium phosphate-IIb transporter protein and subsequently in the formation of microliths due to a reduced phosphate clearance [38]. By direct DNA sequencing, more and more gene variants are being identified [39].
Affected people are often only mildly symptomatic, with dyspnoea, dry cough, chest pain and asthenia [40]. Occasionally, the diagnosis is made incidentally [41]. In more severe cases, cyanosis and clubbing develop, and pneumothoraces may occur in rare cases [40]. Extrapulmonary manifestations includes nephrocalcinosis or nephrolithiasis, and calcifications of the lumbar sympathetic chain, pericardium, aortic and mitral valves, and testicles [42].
While first microliths most probably develop in childhood, the first symptoms occur only decades later [38]. In advanced stages, a decreased transfer factor and a restrictive pattern as well as impaired perfusion and gas exchange develop. This leads to hypoxaemia and, with further progression, pulmonary hypertension and right heart failure [40]. Progression is common, although the rate of disease progression varies considerably [37].
Diagnosis
High-resolution CT (HRCT) shows the pathognomonic sandstorm pattern with small, calcified nodules, areas of ground-glass opacities and consolidation, beginning in the lower lobes (figure S2) [43]. Additionally, calcifications are found alongside interlobular septa, bronchovascular bundles and pleura. Subpleural cysts may also occur in the lung periphery and can cause pneumothorax [44]. When typical, HRCT findings may be sufficient to establish the diagnosis, especially when a family history is present [45].
The diagnosis can be confirmed by the detection of microliths [5] of 50–1000 µm in size [46]. They can be detected in sputum, bronchial wash and BAL [5]. Also, they are detectable in biopsies, where they are PAS-positive, onion-shell-like calcium deposits in the complete alveolar space [47]. Molecular analysis is not mandatory for the diagnostic process, but may play a role once targeted treatment is available [5].
Treatment
To date, most therapeutic approaches to progressive PAM have proven unsuccessful. Attempts were made to remove the microliths by repeated WLL. However, this proved to be ineffective, as were systemic corticosteroids [48]. Sodium etidronate inhibits the formation of hydroxyapatite crystals, but mixed results were reported [49]. The only remaining option therefore is lung transplantation [37] with a favourable long-term outcome [50]. Additionally, supportive therapy should be given, e.g. vaccination and oxygen therapy when needed [51].
PPFE
Introduction
PPFE [52] is a rare form of interstitial pneumonia [53, 54]. It is characterised by upper lobe and subpleural fibrosis [53], involving both the visceral pleura and the subjacent subpleural lung parenchyma, and comprising prominent elastosis of the alveolar walls and fibrous thickening of the visceral pleura [55–57].
Epidemiology
Although rare [58], PPFE is now diagnosed regularly in ILD referral centres. In retrospective studies, PPFE was the main pattern radiologically in 7.7% of idiopathic interstitial pneumonias [59], 25% of fibrotic ILDs registered for lung transplantation [60], 0.28–1.9% of lung transplant recipients [61, 62] and 7.5% of haematopoietic stem cell transplantation recipients [62].
Aetiology and disease associations
PPFE can be idiopathic or secondary to lung or liver transplantation, haematopoietic stem cell transplantation, alkylating drugs, radiation therapy, connective tissue diseases, fibrotic hypersensitivity pneumonia, IPF and other idiopathic interstitial pneumonias, environmental exposures, telomere-related gene mutations, and recurrent respiratory infections. In addition, PPFE-like features can be present in various fibrotic ILDs, including IPF [63–67], fibrotic hypersensitivity pneumonitis [55, 68], systemic sclerosis-associated ILD [69] and rheumatoid arthritis-ILD [70], and are associated with a poor outcome.
Clinical presentation and lung function
PPFE predominates in females, aged 30–60 years, with no link to cigarette smoking. Onset of disease is insidious [54, 56, 71–74], with progressive shortness of breath, dry cough, basolateral pleural chest pain, alteration of general status, weight loss and eventually cachexia [75]. Fine crackles on chest auscultation are present in ∼40% of patients, finger clubbing in 15–20% [75] and platythorax, i.e. anteroposterior flattening of the thorax, in ∼50% [75, 76]. Lung volumes are more severely altered than diffusion capacity for carbon monoxide [75, 76]. A pneumothorax or pneumomediastinum can be inaugural [77], be secondary to lung biopsies [57] and eventually occurs in 25–60% of patients [73, 78]. Pneumothoraces recur in more than half of patients; however, only a minority require chest drainage, which is often complicated by persistent air leak [78].
Diagnosis
Chest CT shows volume loss and dense pleural and subpleural consolidation coexisting with reticulation, architectural distortion and traction bronchiectasis, predominantly in the upper lobes [56, 77–81] (figure 2). PPFE is often more than 5 mm thick and is generally progressive [56]. A pattern of PPFE in the upper lung zones can be associated with a usual interstitial pneumonia (UIP) pattern in the lung bases [63–67].

Computed tomography scan of the chest (parenchymal window) in a 23-year-old female patient with pleuroparenchymal fibroelastosis, demonstrating pleural thickening and subpleural consolidation in the upper lung zones, together with left-sided spontaneous pneumothorax.
Although the diagnosis in theory requires histopathology [82], the risk of pleural complications means that the use of video-assisted thoracoscopic lung biopsy is generally discouraged. In one small series, a majority of patients meeting radiologic criteria for PPFE were histologically confirmed as having PPFE [63]. When performed, the lung biopsy demonstrates subpleural intra-alveolar fibrosis and elastosis, pleural thickening, and prominent deposit of elastic fibre dense collagen [53, 55, 56, 75]. Forceps transbronchial biopsies, transbronchial cryobiopsy and transthoracic core lung biopsy have been successfully used to diagnose PPFE, but with an unclear benefit/risk ratio [83].
Diagnostic criteria were proposed, based on both chest CT and histopathology [56], or on chest CT features and radiologic confirmation of disease progression [75]. The latter are increasingly used to support the diagnosis of PPFE in multidisciplinary discussions.
Outcome
Retrospective series found a heterogeneous disease course, with a median survival time of 35–96 months [59, 75, 76]. The main causes of death are hypercapnic chronic respiratory failure, acute exacerbation of pulmonary fibrosis, cachexia, aspiration pneumonia and pulmonary embolism [59, 75].
Management
Experience with pulmonary rehabilitation [75] and lung transplantation [84] is still limited. Lung transplantation is the only treatment that can cure PPFE [85]. Drug therapy has not been evaluated prospectively. Patients often receive low-dose glucocorticoids; however, higher doses of corticosteroids and second-line immunosuppressive therapy is discouraged due to an increased risk of infections [4]. Data regarding the antifibrotic agents nintedanib and pirfenidone are still preliminary, with conflicting results in two retrospective series for nintedanib [86, 87] and controversial results for pirfenidone [4]. However, in PPFE cases with associated fibrotic progressive ILD, the use of antifibrotics can be considered.
ILD in selected congenital syndromes
Dyskeratosis congenita and telomerase-associated pulmonary fibrosis
Introduction
Telomerase is an enzyme complex composed of the telomerase reverse transcriptase, the telomerase RNA component, dyskerin and other stabilising proteins that help maintain telomere length during mitosis [88]. Dyskeratosis congenita (DC) and telomere biology disorders (TBDs) are multisystem diseases associated with mutations in 16 telomere-related genes [89], with overlapping features including cutaneous abnormalities, premature hair greying, bone marrow failure, liver disease and pulmonary manifestations, of which pulmonary fibrosis is the most common [90].
Epidemiology and clinical presentation
The prevalence of DC is about one in a million [91]. The cumulative incidence of pulmonary fibrosis over 20 years in DC/TBD has been estimated to ∼20% [92]. Telomere-related mutations and/or shortened telomeres are found in approximately 25% of patients with familial pulmonary fibrosis [93–96]. Additionally, telomere-related mutations account for ∼10% of sporadic cases of pulmonary fibrosis [93, 97, 98] and 12% of rheumatoid arthritis-associated ILD [99]. Transmission depends on the gene involved. The shortening of telomeres beyond a critical threshold results in cell senescence or apoptosis [100], especially in tissues with high replicative rates. Additional aggression (smoking, professional exposure) is found in about 70% of patients and is likely to increase the risk of ILD [101, 102].
The clinical spectrum of DC/TBD is broad and ranges from classic dyskeratosis congenita to isolated clinical findings of TBD. The classical clinical triad of DC is lacy reticular hypopigmentation of the skin, dystrophic nails and oral leukoplakia [103]. This well-characterised phenotype mainly affects children and is associated with pulmonary fibrosis in those surviving to the second decade of life. Telomere-related pulmonary fibrosis, however, is more frequent and affects young adults. The following personal or familial findings in a patient referred for pulmonary fibrosis work-up are suggestive of telomere biology disorder: cutaneous, hair and nails abnormalities, cirrhosis or unexplained repeated elevated liver enzymes, bone marrow failure (e.g. myelodysplastic syndromes- features) or milder haematological abnormalities, and head and/or neck cancer [91].
Radiological findings
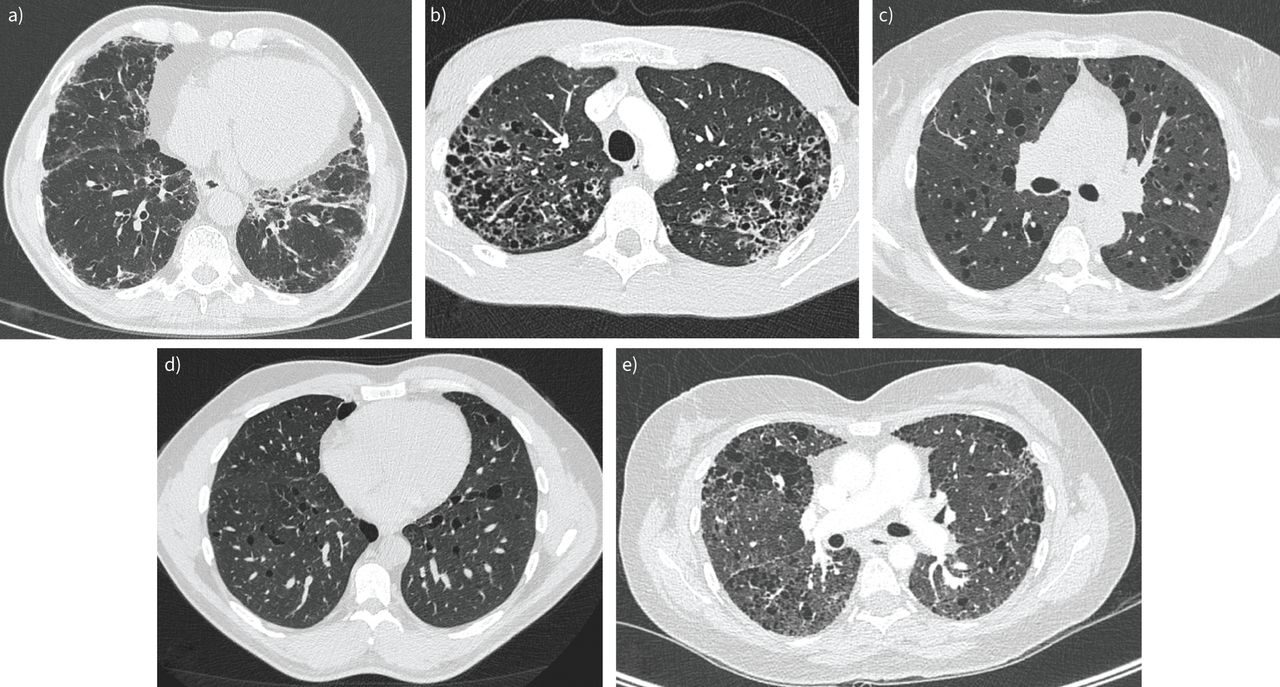
A UIP pattern is seen in half of the patients. In IPF patients with telomere-related gene mutation, atypical features such as air trapping and consolidations may be present (figure 3a) [102, 104, 105]. IPF is the most common diagnosis in approximately half of patients, followed by unclassifiable fibrosis (around 20%) and chronic hypersensitivity pneumonitis (around 10%) [104, 105].
{kind=link}
{kind=link}
{kind=link}
Axial high-resolution computed tomography of different interstitial lung diseases. a) Probable usual interstitial pneumonia pattern in a patient with a heterozygous TERC mutation. b) Bilateral irregular nodular and cystic lesions with upper lobe predominance in a patient with pulmonary Langerhans cell histiocytosis. c) Multiple bilateral centimetric thin-walled cysts in a patient with sporadic lymphangioleiomyomatosis. d) Lower lobe predominant distribution of bilateral thin-walled cysts in a patient with Birt–Hogg–Dubé syndrome. e) Unclassifiable pulmonary fibrosis with microcystic destruction in a patient with a heterozygous SFTPC I73T mutation.
Disease course
Despite the wide range of ILD subtypes associated with telomere-related mutations, the disease course is almost always progressive and comparable to or worse than IPF, regardless of the pattern of lung disease [104]. Particular attention should be paid to extra-respiratory surveillance of these patients to detect other potential telomere-related manifestations (cytopenia, liver disease, cancers).
Diagnosis
Genetic testing is warranted in patients with familial pulmonary fibrosis or sporadic pulmonary fibrosis with familial or personal findings suggestive of TBD. Indication of lung transplantation for pulmonary fibrosis should also prompt testing for telomere-related mutations [106].
Treatment
In patients with progressive fibrotic ILD, antifibrotic therapy should be implemented. Danazol, a synthetic androgen, is currently being evaluated in short telomere-associated pulmonary fibrosis in a phase 2 randomised trial [107]. Patients with telomere-related gene disorders and rapidly progressive lung disease should be referred early to lung transplant centres; however, particular attention must be paid to immunosuppressant therapy with respect to bone marrow reserve. In patients with a complete DC phenotype, all reported lung transplantation cases were preceded by bone marrow transplantation [108]. Gene therapy might be a future option [109].
Surfactant-associated pulmonary fibrosis
Introduction
Surfactant-related disorders range from fatal neonatal respiratory distress syndrome to familial ILDs in children and adults. Surfactant-related genes involved in adult-onset pulmonary fibrosis code for SP-A1 and SP-A2, SP-C, ATP-binding cassette transporter A3 (ABCA3) and thyroid transcription factor NK2 homeobox 1 (NKX2-1) [110, 111]. Mutations in SFTPA1/2 and NKX2-1 are also associated with lung cancer [112].
Epidemiology and clinical presentation
Mutations in surfactant-related genes account for approximately 3–5% of familial pulmonary fibrosis cases [113]. Inheritance is autosomal dominant with incomplete penetrance for SFTPC and SFTPA1/2 mutations and autosomal recessive for ABCA3 mutations.
Depending on the protein domain involved, mutations in SFTPC promote ILD and pulmonary fibrosis by impairment of autophagy and phospholipids recycling, increased cellular sensitivity to injury or cytotoxicity resulting from aggregation of misfolded protein [114–116]. Mutations in SFTPA1 or SFTPA2 result in reduced protein secretion, thereby impairing normal surfactant function and disrupting the lung inflammation modulation ability of SP-A [112, 117]. ABCA3 mutations also impair normal surfactant function by preventing the formation of lamellar bodies [118].
Phenotypic heterogeneity is considerable in surfactant-related pulmonary fibrosis, ranging from asymptomatic patients to end-stage respiratory insufficiency. Severe pulmonary hypertension may develop. Age of onset varies from infancy to late adulthood. Neonatal diagnosis is frequent in homozygous ABCA3 mutation carriers but is uncommon in SFTPC and SFTPA1/2 mutation carriers [112, 119, 120].
Radiological findings
HRCT features reflect the clinical heterogeneity, with no clear genotype–phenotype correlation. The most frequent pattern seen in SFTPC or ABCA3-related ILD is the association of diffuse ground-glass opacities with cysts and septal thickening (figure 3e), with honeycombing later in the course of the disease. Combined pulmonary fibrosis with emphysema is also described [112, 121–124].
Diagnosis
Occurrence of pulmonary fibrosis in a familial context or before 50 years old should prompt genetic testing for telomerase and surfactant-related genes [113]. The suspected mode of transmission helps to tailor genetic analysis.
HPS
Introduction
HPS is a group of autosomal recessive disorders combining bleeding diathesis and oculocutaneous albinism, caused by mutations in 11 genes [126]. HPS can also be associated with granulomatous colitis [127] and pulmonary fibrosis, depending on the subtypes.
Epidemiology and clinical presentation
HPS worldwide prevalence is estimated at one to nine cases per million individuals, with the highest worldwide prevalence of 1:1800 in north-western Puerto Rico [128]. Only patients with HPS types 1, 2 and 4 are prone to ILD [127]. All patients with HPS type 1 will develop pulmonary fibrosis [129].
The mutations in HPS-related genes cause dysfunction in lysosome-related organelles. Putative mechanisms of fibrogenesis include lysosomal accumulation of ceroid material [130], autophagy [131], impaired surfactant trafficking and endoplasmic reticulum stress [132].
Ocular and cutaneous manifestations include variable pigmentation of hair and skin, horizontal nystagmus, and impaired visual acuity [133]. Bleeding symptoms include spontaneous bruising and prolonged minor bleedings (epistaxis, menorrhagia). ILD is often diagnosed during the third and fourth decades of life [127].
Radiologic findings
Features seen on chest CT include septal thickening, diffuse ground-glass opacities, peribronchovascular and pleural thickening. A reticular pattern with subpleural honeycombing may be present in advanced disease [127]. Ground-glass opacities might be the main radiological feature [134].
Diagnosis
The diagnosis of HPS is based on the demonstration of oculocutaneous albinism in the presence of a deficit in platelet storage pool. Genetic testing is used to confirm the diagnosis and subtype [126].
Management
There are no specific therapeutic options for the treatment of HPS-associated pulmonary fibrosis. Two trials of pirfenidone in HPS patients were not conclusive [136–138]. In patients with advanced disease, lung transplantation is the only therapeutic option for eligible patients. HPS-related bleeding diathesis has been considered a contraindication for lung transplantation, but a good outcome has been reported with appropriate bleeding diathesis prophylaxis [139]. Otherwise, management is based on palliative care [140].
Other rare diffuse lung diseases of genetic origin
The genetic cause is still unknown in 60% of cases of familial pulmonary fibrosis. Several diffuse lung diseases linked with genetic defects not described in detail in this review are briefly summarised in table 2.
Overview over other rare diffuse lung diseases of genetic origin
Multiple cystic lung diseases
Lymphangioleiomyomatosis (LAM)
LAM is a very rare multisystem disease that primarily affects women, either sporadically or in association with tuberous sclerosis complex (TSC) [147, 148]. LAM is characterised by the presence of abnormal smooth muscle-like LAM cells from an unknown source and infiltrating the tissues. Beside lung manifestations, LAM is associated with angiomyolipomas, lymphangioleiomyomas, chylous effusions [149] and, in a TSC setting, with epilepsy and cutaneous abnormalities [147].
Epidemiology and clinical presentation
Prevalence of LAM has been estimated as five cases per million women, with great variations suggesting that this number is underestimated [150]. LAM is sporadic in two-thirds of cases. In patients with TSC, prevalence of LAM increases with age, reaching 81% at the age of 40 [147].
Both TSC-LAM and sporadic LAM are caused by mutations in TSC1 and TSC2 [147, 151] resulting in uncontrolled cellular proliferation, expression of vascular endothelial growth factor (VEGF)-A, VEGF-C and VEGF-D, angiogenesis stimulation, immune evasion, and cell survival [149, 152].
LAM typically occurs in women of childbearing age. Respiratory symptoms are nonspecific: dyspnoea, cough, wheezing and haemoptysis. Pleural or peritoneal chylous effusions are seen in 20% and 5% of the cases, respectively [153]. Spontaneous pneumothorax is the presenting manifestation in 24–36% of patients. Around 40% of patients present renal angiomyolipomas, more frequently in TSC [153]. Pulmonary function testing often reveals airway obstruction [153, 154].
Radiological findings
On HRCT, LAM is characterised by diffuse cystic changes without predilection in distribution. Cysts are thin-walled and of intermediate size (figure 3c). Additional findings include ground-glass opacities, interlobular septa thickening, pneumothoraces and pleural effusion [155].
Disease course
Women before menopause experience a more marked lung function decline and exacerbations can occur during pregnancy or oestrogen supplementation [156], whereas the disease tends to abate after menopause. The 10- and 20-year transplantation-free survival rates are 85% and 64% respectively [157].
Diagnosis
Diagnosis is achieved in the setting of characteristic or compatible HRCT pattern with a combination of other characteristics, including angiomyolipoma, lymphangioleiomyoma, confirmed or probable TSC, or chylous effusion or a VEGF-D ≥800 pg·mL−1 or consistent histology [158, 159].
Management
Sirolimus, a mammalian target of rapamycin (mTOR) inhibitor, has been shown to effectively stabilise lung function and alleviate angiomyolipomas [160–162]. Everolimus, another mTOR inhibitor, seems to be similarly effective [163]. Some studies are ongoing with several drugs for treatment of LAM. Given the high rate of pneumothorax recurrence, pleurodesis should be considered after the first episode. In end-stage respiratory failure, lung transplantation is a viable option with a reported median survival of 12 years [164]. Previous pleurodesis is not contra-indicative of lung transplantation.
Birt–Hogg–Dubé syndrome (BHDS)
BHDS caused by germline mutations in FLCN, the folliculin gene [165], is associated with lung cysts, spontaneous pneumothoraces, renal tumours and typical skin lesions named fibrofolliculomas.
Epidemiology and clinical presentation
The prevalence of BHDS in the general population has been estimated at around two cases per million based on epidemiological data inferred from spontaneous pneumothoraces [166].
The exact mechanism of renal tumorigenesis and lung cysts formation in BHDS is unknown, but a tumour-suppressor role of folliculin is postulated through the Akt-mTOR pathway [165]. In most renal tumours from BHDS patients, additional FLCN mutations are found, fitting the two-hits model of oncogenesis [167].
Lung cysts are present in around 85% of patients, resulting in 30–70% of them in spontaneous pneumothoraces, which are often the presenting manifestation [168]. Most patients have normal lung function. The classical fibrofolliculomas are hair follicle tumours, presenting as white or skin-coloured papules of the face and upper torso. Lung cysts are present from the second decade of life, the majority of pneumothoraces occur between the ages of 40 and 50 years, and renal tumours are diagnosed after 40 [168].
Diagnosis
Diagnosis of BHDS is made in patients with at least five fibrofolliculomas or carrying a heterozygous pathogenic mutation in FLCN. The diagnosis can be made if two of the following criteria are present: multiple lung cysts compatible with BHD, renal cancer before 50 years old, multifocal or bilateral renal cancer, hybrid chromophobe-oncocytic renal cancer, or a first relative with BHDS [165].
Management
There is no specific treatment of BHDS. The management of pneumothorax is similar to other pneumothorax patients. A screening and follow-up for renal cancer is suggested [170]. Fibrofolliculomas can be treated by laser, surgery or dermal sirolimus application, but recurrence is frequent [171].
Pulmonary Langerhans cell histiocytosis (PLCH)
Langerhans cells are a subset of dendritic cells, localised in the epiderma and the mucosal epithelium (including the airways epithelium), characterised by cluster of differentiation (CD) 1a and CD207 (“Langerin”) expression [172]. In PLCH, accumulation of CD1a+ cells in the lungs causes inflammatory cystic-nodular pulmonary parenchyma destruction in predisposed smokers [173].
Epidemiology and clinical presentation
One survey-based study in Japan reported a prevalence of 0.27 in males and 0.07 in females per 100 000 people [174], which is probably underestimated. The strongest risk and likely causal factor is current or former tobacco smoking [173, 175].
Somatic activating mutations in genes of the mitogen-activated protein kinase pathway are found at the inception of the disease [176, 177]. The mutated CD1a+ dendritic cells exhibit resistance to apoptosis and a decreased tendency to exit the lung through lymph nodes, resulting in the formation of cellular nodules and the subsequent centrilobular fibrotic lung remodelling [178]. Cigarette smoke probably acts as a “second hit”.
PLCH often presents in the fourth decade of life [173]. Symptoms include cough, dyspnoea, chest pain, fever and general malaise, but up to 50% of patients are asymptomatic [174]. Approximately 20% of patients have extrapulmonary manifestations, most frequently bone and pituitary gland lesions [173, 174]. The most frequent anomaly on pulmonary function tests is reduced diffusing capacity [179].
Radiological findings
The typical aspect on HRCT consists of bronchiolocentric micronodular/nodular and cystic lesions with parenchymal destruction, of predominant upper and middle lobes distribution with typical sparing of costophrenic angles [180–182]. Depending on the stage of PLCH, the octopus sign (central scar with septal strands and associated airspace enlargement) can be identified [183]. Cysts can be of various shapes (round, bilobed, clover-leafed, irregular or “bizarre”) (figure 3b).
Disease course
Most of the cases experience regression with smoking discontinuation but in around 20–30% of the cases the disease progresses to end-stage lung disease [184]. Between 12–32% of patients will present pneumothorax in their lifetime [185]. A prospective series followed newly diagnosed patients with PLCH for 2 years and found that 38% presented lung function decline, with predictors of deterioration being smoking status and baseline PaO2 [175]. 10-year survival has been estimated at 93% [179].
Diagnosis
A definite diagnosis requires confirmation either by BAL or sometimes histology, but a high-confidence diagnosis can often be made with a combination of a typical HRCT pattern and suggestive history, especially in smokers [186].
Treatment
Smoking cessation alone can be sufficient to obtain stabilisation or regression of PLCH [187], but tobacco and marijuana weaning are sometimes temporary and are insufficient in approximately one-third of cases. Pharmacological treatment with cladribine has been shown to be effective [188] and BRAF kinase/MEK inhibition represents promising leads in case for specific mutations [189]. In advanced cases, lung transplantation is effective, but recurrence can occur [190].
Conclusion
Many rare and ultrarare ILDs are likely underdiagnosed. Increasing insights into their pathobiology, genetics and disease behaviour have led to the development of better treatment options, with very effective targeted therapies for some of the diseases, whilst in others transplant or palliative care may be the only remaining option. To diagnose, optimally treat and support patients with rare ILDs as well as advance the field, early consultation with or referral to expert centres is advised. The organisation of these expert centres for rare ILDs within the European Reference Network on rare respiratory diseases (ERN-LUNG) further stimulates clinical and research collaboration to advance the care for patients with rare ILDs across Europe.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary figures S1 and S2 ERR-0161-2022.supplement
Footnotes
Provenance: Commissioned article, peer reviewed.
Previous articles in this series: No. 1: Simonneau G, Fadel E, Vonk Noordegraaf A, et al. Highlights from the International Chronic Thromboembolic Pulmonary Hypertension Congress 2021. Eur Respir Rev 2023; 32: 220132.
Number 2 in the Series “The world of rare lung diseases” Edited by Michael Kreuter, Marc Humbert, Thomas Wagner and Marlies Wijsenbeek
This article has an editorial commentary: https://doi.org/10.1183/16000617.0006-2023
Conflict of interest: K. Buschulte has received payment or honoraria for lectures, presentations, speakers’ bureaus, manuscript writing or educational events from BI.
Conflict of interest: V. Cottin has received an unrestricted grant to his institution from Boehringer Ingelheim; consulting fees from Boehringer Ingelheim, Roche, Shionogi, RedX, Pure Tech, Celgene/BMS, AstraZeneca, CSL Behring, Sanofi, United Therapeutics/Ferrer and Pliant; fees for lectures from Boehringer Ingelheim and Roche; support for attending meetings from Boehringer Ingelheim and Roche; reports participation on a Data Safety Monitoring board for Galapagos and Galecto; and participated in an Adjudication committee for Fibrogen.
Conflict of interest: M. Wijsenbeek has received payment for a DSMB from savarapharma.
Conflict of interest: M. Kreuter has received grants or contracts, consulting fees and payment or honoraria for lectures, presentations, speakers’ bureaus, manuscript writing or educational events from BI and Roche; and has leadership or fiduciary roles at ERS, EU IPFF and DGP.
Conflict of interest: R. Diesler has received support for attending meetings and/or travel from Laidet Medical/Asdia and Vitalaire; and has other financial or non-financial interests (conference organisation) with Laidet Medical/Asdia, Vitalaire, ALLP, Agiradom and Lowenstein.
- Received September 8, 2022.
- Accepted December 21, 2022.
- Copyright ©The authors 2023
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References










