





Abstract
The lungs face ongoing chemical, mechanical, biological, immunological and xenobiotic stresses over a lifetime. Advancing age progressively impairs lung function. Autophagy is a “housekeeping” survival strategy involved in numerous physiological and pathological processes in all eukaryotic cells. Autophagic activity decreases with age in several species, whereas its basic activity extends throughout the lifespan of most animals. Dysregulation of autophagy has been proven to be closely related to the pathogenesis of several ageing-related pulmonary diseases. This review summarises the role of autophagy in the pathogenesis of pulmonary diseases associated with or occurring in the context of ageing, including acute lung injury, chronic obstructive pulmonary disease, asthma and pulmonary fibrosis, and describes its potential as a therapeutic target.
Abstract
Autophagy is a “housekeeping” survival strategy and its activity decreases with age. Manoeuvring autophagy can be a potential therapeutic target in ageing-related pulmonary diseases. https://bit.ly/3URj2bN
Introduction
The lungs are not only the primary location where gas exchange occurs in mammals but also have the largest epithelial surface area in the body; therefore, the lungs represent a unique interface with the outside environment and face ongoing chemical, mechanical, biological, immunological and xenobiotic stresses over a lifetime [1, 2].
Over the last century, due to improvements in living conditions and the continuous development of medical technology, the human lifespan has increased dramatically, and the population is ageing at a rate never seen before in human history [2, 3]. Age-related ailments represent a formidable global socioeconomic burden and a significant healthcare challenge [4, 5]. Advancing age progressively impairs lung function in otherwise healthy individuals; lung-resident cells rely on robust stress response pathways to stave off cumulative damage, yet in an ageing lung, the homeostatic control of wound healing following stress has an increased likelihood of being perturbed, elevating susceptibility to diseases. Therefore, many lung diseases are more prevalent and lethal in the elderly. Specifically, the recent coronavirus disease 2019 (COVID-19) pandemic, which further contributes to the global impact of lung diseases, highlights the increased susceptibility of the elderly to acute respiratory distress syndrome (ARDS) and subsequent fibrosis [6] and underscores whether there are age-related molecular determinants that can be targeted for therapeutic purposes to mitigate morbidity and mortality in elderly populations [7]. At the molecular and cellular levels, the nine hallmarks of ageing (genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion and altered intercellular communication) are listed by López-Otín et al. [8] and are implicated in the pathogenesis of ageing-related lung diseases [9–11].
Autophagy is a “housekeeping” survival strategy involved in numerous physiological and pathological processes in all eukaryotic cells [12]. Autophagic activity decreases with age in several species, whereas the basic activity of autophagy extends throughout the lifespan in most animals, indicating that it is one of the convergent mechanisms of several longevity pathways [13]. Moreover, dysregulation of autophagy has been proven to be closely related to the pathogenesis of several pulmonary diseases.
In this review, we outline the mechanism of autophagy, focusing on its fundamental mechanisms in ageing and age-related pulmonary diseases, and discuss the therapeutic potential of regulating autophagy.
Overview of autophagy
Three types of autophagy have been distinguished based on their differing mechanisms of cargo sequestration. In this study, we discuss the most studied form of autophagy: macroautophagy (hereafter simply referred to as autophagy). Autophagy involves the formation of autophagosomes (double-membraned vesicles) and their fusion with endosomes or lysosomes to form amphisomes or autolysosomes. Both the formation and turnover of the autophagosome involve evolutionarily conserved genes: autophagy-related (Atg) genes [14]. Autophagy is highly dynamic, and its process is typically divided into distinct stages: initiation, elongation/closure and autophagosome–lysosome fusion (figure 1).

Illustration of autophagy. Autophagy involves the formation of autophagosomes and their fusion with lysosomes to form autolysosomes. The process is typically divided into distinct stages: initiation, elongation/closure and autophagosome–lysosome fusion. Initiation begins with activation of the complex. Unc-51-like autophagy activating kinase 1 (ULK1) and Atg13 are key to the ULK1 complex and are further supported by Atg101 and FAK family-interacting protein of 200 kDa (FIP200). Mammalian target of rapamycin complex 1 (mTORC1) binds to ULK1 and inhibits the ULK1 complex, and 5ʹ AMP-activated protein kinase (AMPK) phosphorylates mTORC1, resulting in the dissociation of mTORC1 from ULK. The ULK1 complex activates a class III PI3K complex consisting of VPS34, VPS15, Beclin-1 and Atg14L and activates the molecule in BECN1-regulated autophagy protein 1 (AMBRA1). The phagophore elongates and encloses to a double-membrane autophagosome. This step is tightly regulated via the ubiquitin-like conjugation systems. For example, the Atg12–Atg5:Atg16L1 complex conjugates phosphoethanolamine to LC3 to LC3-II, and LC3-II promotes substrate uptake upon binding to different receptors, such as p62. The autophagosome fuses with a lysosome to form an autolysosome for degradation. The inner content is released into the lysosome/autolysosome and is degraded by lysosomal hydrolases. The SNARE-like proteins may play critical roles in autophagosome–lysosome degradation. LAMP1: lysosomal-associated membrane protein 1.
Based on cargo specificity and delivery mechanisms, autophagy can be divided into two types: nonselective autophagy and selective autophagy (including mitophagy, pexophagy, endoplasmic reticulum (ER)-phagy, ribophagy and lipophagy).
Physiological role of autophagy and impact of autophagy on ageing and ageing-related pulmonary dysfunction
Role of autophagy in biology
Autophagy modulates various physiological processes in cells and plays a fundamental role in cellular, tissue and organismal homeostasis [12]. Therefore, the occurrence of the “diseased” state may be associated with autophagy dysregulation due to alterations in the central aspects of multicellular organism biology [15].
Autophagy is a particularly important regulatory mechanism in the lungs. Autophagy is abundant in lung epithelial cells. Epithelial autophagy is activated in the developing mouse lung through 5ʹ AMP-activated protein kinase (AMPK) activation, and the inhibition of AMPK-mediated autophagy reduced lung branching in vitro. The conditional deletion of Beclin-1 in mouse lung epithelial cells, at either early or late gestation, resulted in lethal respiratory distress at birth or shortly after birth [16].
Role of autophagy in ageing and lifespan
Autophagy activation has been proven to extend animal lifespan; it decreases with age in numerous species [17, 18]. The transcriptional and epigenetic regulation of Atg genes impacts the physiology and lifespan of animals [19, 20]. For example, mutations or loss of function in Atg1, Atg7, Atg18 and Bec-1 reduced the lifespan of Caenorhabditis elegans [21]. In contrast, the overexpression of Atg5 extended the lifespan of mice [22]. However, several investigations suggested that autophagy can be harmful under certain circumstances and in some models. For example, moderate overexpression of Atg1 substantially extends the lifespan of Drosophila; however, strong expression is toxic [23]. These observations may reflect the complexity of autophagy in ageing modulation. Nonetheless, we can infer that the maintenance of functional autophagy is essential for organismal ageing and that dysregulation of autophagy, whether insufficient or excessive, contributes to cellular deficits and functional organismal decline.
Crosstalk between autophagy, ER stress/unfolded protein response (UPR) and apoptosis
Besides autophagy, ER stress/UPR and apoptosis are two fundamental biological processes essential for manifold cellular functions in health and disease [24]. Alteration in the expression of apoptosis, autophagy and UPR markers is correlated with lung function in lung tissue [24].
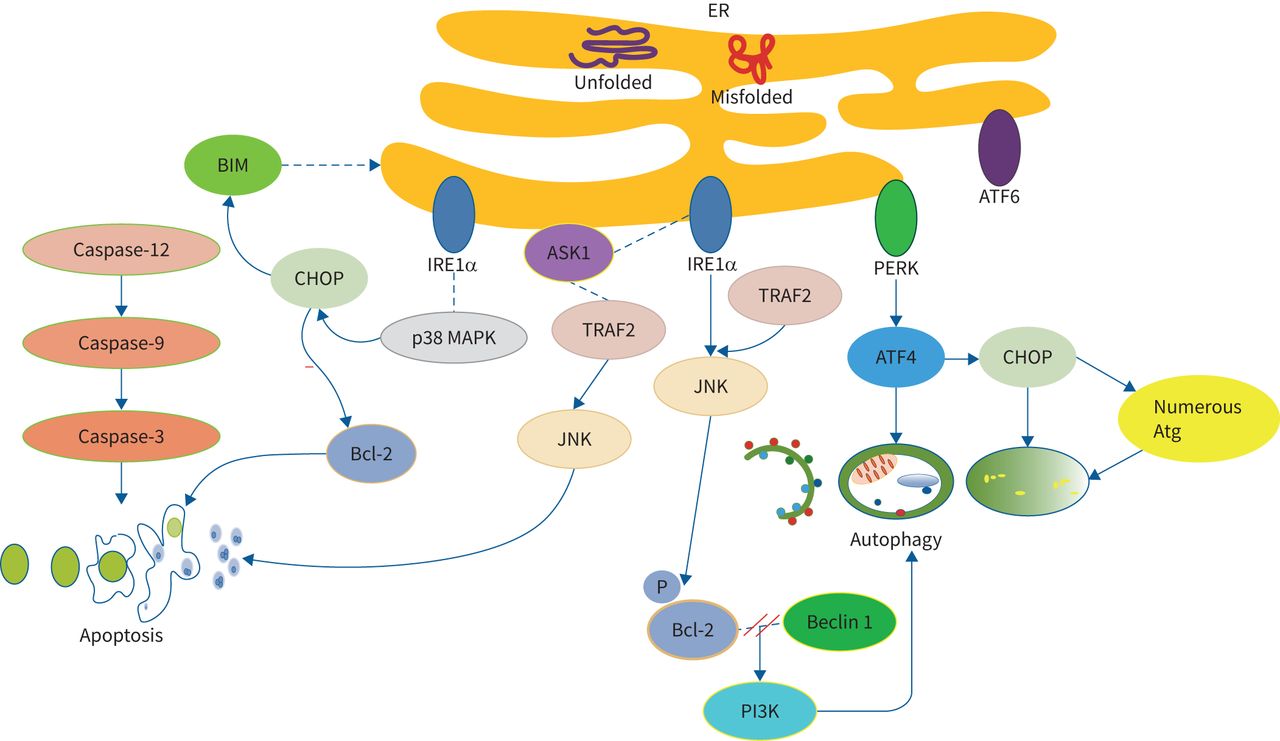
The UPR system is controlled by three ER transmembrane proteins and 78 kDa glucose-regulated protein. Transmembrane proteins include inositol-requiring enzyme 1α (IRE1α), protein kinase RNA-like ER kinase (PERK) and acting transcription factor (ATF) 6 [25]. Autophagy is activated with persistent ER stress to promote cell survival. ATF4 and CCAAT/enhancer-binding protein (C/EBP) and homologous protein (CHOP) (both activated by PERK) are mostly related with cellular death pathways upon overexpression. Autophagy is transcriptionally regulated by ATF4 and CHOP and can oppose terminal UPR and ATF4 [26]. CHOP has also been identified in the regulation of numerous Atg genes [27]. ER stress also activates c-Jun NH2-terminal kinase (JNK) via the interaction of IRE1α and TNF receptor-associated factor 2 (TRAF2), ultimately phosphorylating Bcl-2 and leading to dissociation of Bcl-2 and Beclin-1 proteins. This enables activation of the PI3K complex and initiates autophagy. ER stress-induced activation of apoptosis requires IRE1α, which forms a complex with apoptosis signal-regulating kinase 1 (ASK1) and stimulates its downstream target, JNK, via binding to TRAF2. In addition, ER stress induces apoptosis via a caspase-dependent pathway. It leads to the translocation of the BH3-only protein, Bcl-2-like protein 11 (BIM), to the ER, leading to caspase-12 activation. Caspase-12 activates caspase-9, leading to downstream caspase-3 activation in this cascade, finally triggering apoptosis. Additionally, IRE1α can cause phosphorylation and activation of CHOP through binding and activation of p38 mitogen-activated protein kinase. CHOP downregulates Bcl-2 and upregulates the transcription of BIM, eventually causing the downstream initiation of an apoptotic cascade [28].
The crosstalk of autophagy with ER stress and apoptosis is illustrated in figure 2.

The crosstalk of autophagy with endoplasmic reticulum (ER) stress and apoptosis. ER stress activates autophagy to promote cell survival. Autophagy is transcriptionally regulated by acting transcription factor 4 (ATF4) and CCAAT/enhancer-binding protein homologous protein (CHOP) and can oppose terminal unfolded protein response and ATF4. CHOP regulates numerous Atg. ER stress activates c-Jun NH2-terminal kinase (JNK) via the interaction of inositol-requiring enzyme 1α (IRE1α) and TNF receptor-associated factor 2 (TRAF2), ultimately phosphorylating Bcl-2 and leading to dissociation of Bcl-2 and Beclin-1 proteins. This enables the activation of the PI3K complex and initiates autophagy. ER-stress-induced activation of apoptosis requires IRE1α. IRE1α forms a complex with apoptosis signal-regulating kinase 1 (ASK1) and stimulates its downstream target, JNK, via binding to TRAF2. ER-stress-induced apoptosis via a caspase-dependent pathway. ER stress leads to the translocation of Bcl-2-like protein 11 (BIM) to the ER, leading to caspase-12 activation. Caspase-12 activates caspase-9, leading to downstream caspase-3 activation in this cascade, finally triggering apoptosis. Additionally, IRE1α causes the phosphorylation and activation of CHOP through binding and activation of p38 mitogen-activated protein kinase (MAPK). CHOP downregulates Bcl-2 and upregulates the transcription of BIM, eventually causing the downstream initiation of an apoptotic cascade. PERK: protein kinase RNA-like ER kinase.
Recent reports highlight the importance of interplay between ER stress/UPR, autophagy and apoptosis in lung diseases. For example, mevalonate cascade inhibition, which has been linked with improved lung health, leads to cell death via coordinated apoptosis, autophagy and ER stress [29]. The interplay between these pathways appears to be mainly regulated via autophagy and Bcl-2-family pro-apoptotic proteins [30]. In COPD, the chaperone and master regulator of the UPR-BiP may be upregulated in bronchoalveolar lavage fluid and lung samples taken from smokers. The expression levels of phospho-translation initiation factor 2α (eIF2α) and CHOP correlate with the severity of airway obstruction, and these increments are associated with stress-induced increases in caspase-3 and -7. COPD patients also show a significant elevation in autophagic proteins. Activation of the PERK-eIF2α axis is critical for the autophagy activity associated with ER stress. Further, PERK activates autophagy by inhibiting Akt/ATF4-mediated induction of Atg [28]. The upregulation of autophagy is linked with airway remodelling in asthma [31], as autophagy facilitates the extracellular matrix deposition and fibrosis in asthmatic airway remodelling [30]. ER stress and UPR activation in airway epithelial cells (AECs) adversely affects asthma, and acute and prolonged activation of UPR proteins can lead to the development of an allergic airway inflammation. UPR simultaneously mediates apoptosis in neutrophil cells via the activation of the CHOP-PERK arm [25]. A recent study showed that the expression of apoptosis, autophagy and UPR markers correlates with lung function deficits in idiopathic pulmonary fibrosis (IPF). The cell stress markers of BiP, X-box binding protein (XBP1), LC3β puncta and cleaved caspase-3 were elevated in IPF lungs compared to non-IPF lungs. However, the mechanisms linking UPR and autophagy in IPF and the imbalance in these cell stress pathways requires further research [24].
Roles of autophagy in ageing-related pulmonary diseases
Acute lung injury (ALI)
The incidence and mortality of ALI and severe ARDS markedly increase with advancing age [32]. ALI is a common and severe clinical syndrome, characterised by noncardiogenic pulmonary oedema, increased alveolar-capillary permeability, neutrophil recruitment and diffused alveolar damage, and is a major cause of acute respiratory failure [33]. The inflammatory response in the lungs with the release of proinflammatory cytokines can be observed in the pathogenesis of ALI [34, 35]. The involved wound repair mechanism, namely, the fibro-proliferative response, if excessive and persistent, will lead to interstitial fibrosis [36, 37].
Evidence proves that autophagy is stimulated in response to various stimuli of ALI, such as bacterial infection, lipopolysaccharide, sepsis, hyperoxia and COVID-19 [1, 38, 39]. The loss of Atg genes, such as Atg7, Atg5 and Atg4B, markedly aggravates the development of ALI in mice [38, 40], indicating that autophagy has protective effects against the initiation and progression of ALI in certain contexts.
COPD
COPD is the fourth leading cause of death worldwide, with increasing prevalence, particularly in the elderly, and ageing hallmarks are prominent features of COPD. COPD is characterised by dysfunctional tissue repair, resulting in (small) airway diseases and emphysema, manifested as persistent respiratory symptoms and airflow limitations [41]. Cigarette smoke extract (CSE) exposure is the most common risk factor for COPD [42, 43]. The pathogenesis of COPD remains incompletely understood; however, increasing evidence has shown that autophagy may be involved in the pathogenesis of COPD. In vitro studies using lower doses of CSE have shown that loss of autophagy enhances smoke-induced epithelial cell senescence, mitochondrial reactive oxygen species (ROS) production, and the accumulation of ubiquitinated proteins along with the accumulation of sequestosome 1 [44–46], suggesting that autophagy plays a protective role in epithelial cells in the pathogenesis of COPD. The chemical activation of autophagy also protects cells in vitro [45] and in vivo from smoke exposure [47]. However, other studies conversely demonstrated that the activation of autophagy, particularly selective autophagy, has harmful effects on epithelial cells in response to smoke [48–51]. CSE increases autophagosomal turnover (flux) and promotes epithelial cell death both in vitro and in vivo [52, 53] and then initiates and exaggerates airway inflammation [54] and mucus hyperproduction [55]. These contradictory results suggest the dual role of autophagy in COPD.
Asthma
Asthma is a complex disorder of the airways involving bronchial hyperreactivity, chronic airway inflammation, mucus overproduction and airway wall remodelling [56, 57]. Ageing-associated change in immune responses facilitates the pathogenesis of asthma in the elderly [58]. Autophagy is mainly involved in the pathogenesis of asthma via the regulation of the body's innate and adaptive immune responses [59]. It is involved in several key processes of asthma pathogenesis, such as airway hyperresponsiveness [60], eosinophilic airway inflammation [61] and airway remodelling [31]. It may also promote interleukin (IL)-18 secretion in response to outdoor allergens in AECs [62]. Bronchial fibroblasts in patients with severe asthma exhibit increased mitophagy and expression of PTEN-inducible putative kinase 1 (PINK1) and Parkin, as well as an increased light chain 3 phosphatidylethanolamine conjugate (LC3-II) expression and a profibrotic phenotype [63]. In vitro human AECs showed that IL-13 stimulates goblet cell formation and mucin 5AC secretion, correlating with the activation of autophagy, manifested by an increase in LC3-II and increased autophagic flux, which can be prevented by Atg5 knockdown [64]. Similarly, Atg5 is correlated with reduced lung function and airway remodelling in patients with severe asthma [65].
IPF
IPF is an interstitial lung disease characterised by massive deposition of the extracellular matrix in the lung interstitium and the irreversible and slowly progressive destruction of lung structure and function [66]. The aetiology of IPF is unknown and ageing is one of the most significant risk factors for IPF. Fibroblast senescence establishes a close link between cellular senescence and IPF [67], senolytics that remove senescent fibroblasts decreased pulmonary fibrosis (PF) in a mouse model of IPF [68, 69].
The induction or enhancement of autophagy has anti-fibrosis effects; autophagy deficiency can promote the deposition of extracellular matrix in lung fibroblasts and accelerate the process of fibrosis [70, 71]. Reduced autophagy can induce epithelial–mesenchymal transition of AECs and contribute to fibrosis via aberrant epithelial–fibroblast crosstalk [72]. However, another study showed that autophagy may also promote the profibrotic effects of transforming growth factor β1 in human lung fibroblasts [73]. Moreover, Akt1-mediated mitophagy contributes to alveolar macrophage apoptosis resistance and PF development [74]. Further studies addressing the regulatory network of autophagy in PF are necessary.
The specific autophagy receptors and markers that change in lung diseases are summarised in table 1.
The specific autophagy receptors and markers that change in lung diseases
The anti-ageing mechanism of autophagy in pulmonary dysfunction
Ageing results in physiological, structural and mechanical changes that diminish lung function. In this condition, insults to the ageing lung are more likely to lead to pathological repair rather than wound resolution and restitution in the lungs. Animal studies proved that older mice are more prone to lung injury; older mice take a longer time to recover than younger mice [75, 76]. Increasing evidence proves that the activity of autophagy is altered in the ageing lung and modulating autophagy may be a promising strategy for treating lung diseases, particularly those in the elderly.
Autophagy maintains protein homeostasis
Loss of proteostasis is a hallmark of ageing [8]. Advanced age itself alone can impair the physiological function of the lung even in the absence of diseases [2]. When the lung faces various biochemical perturbations over a lifetime, it leads to impaired protein homeostasis that urge a robust response to resultant proteotoxic stress. Proteomic analysis of type II AECs isolated from young and old mice revealed a maladaptive collapse of the proteostasis network due to age and the critical role for the co-chaperone adaptive response network in handling chronic misfolded proteins in the ageing healthy lung [2]. Several genetic respiratory diseases are attributable to protein misfolding due to underlying genetic mutations. For example, mutations in the surfactant protein C gene and in the mucin 5B promoter region [77, 78] are related to PF.
Along with the ubiquitin–proteasome system, autophagy is a principal proteolytic system that plays a central role in maintaining cellular proteostasis [8]. The genetic inhibition of core components or regulators of the autophagy machinery markedly accelerates age-related protein aggregation, manifested as shortened lifespan and exacerbated pathological features in worm, fly and animal models of diseases. Disturbance of the interrelationship between proteasomes and autophagy pathways may lead to the accumulation of aberrant proteins and eventually result in pathological conditions in the lungs. For example, smoke-induced aggresome formation contributes to the COPD–emphysema pathogenesis and is related to impaired autophagy; autophagy-inducing drugs markedly decrease aggresome colocalisation and expression [47, 79]. In acute injury, such as chemical lung injury with bleomycin, autophagy decreases with the corresponding elevated levels of oxidised proteins and lipofuscin in response to lung injury in old and middle-aged mice compared with that in younger animals. Older mice with lung injury are characterised by deficient autophagic response and reduced mitophagy [80].
Autophagy modulates stem cell function
Reduction in the regenerative potential of tissues is one of the most obvious characteristics of ageing [8]. In the airways, basal cells (BCs) are multipotent stem cells that form part of the pseudostratified epithelium of the bronchi and trachea. Type II AECs are the main progenitor cell population in the alveolar parenchyma. Type II AECs regenerate after injury by proliferating and differentiating into type I cells, which are critical to gas exchange [81]. Age can cause both quantitative and qualitative changes in various progenitor populations in the lungs. For example, BCs and club cells decrease in number with age, whereas type II AECs do not decrease in quantity but exhibit reductions in self-renewal and differentiation capacity [82–84.]
Dysregulation of stem cell maintenance has been implicated in the pathogenesis of chronic lung diseases; for example, chronic exposure to cigarette smoke (CS) induces a proinflammatory microenvironment that distorts the BC transcriptome, generating pathological epithelial phenotypes [85]. The eventual progenitor exhaustion is responsible for the pathogenesis of COPD [86]. The mechanism through which ageing reshapes the lung stem niche still needs to be elucidated and declined autophagy may be linked to stem cell function reduction during the ageing process. CS-induced senescence in type II AECs is related to decreased autophagy, which enhances p21-mediated cellular senescence [87] and promotes apoptosis in type II AECs [88], while both apoptosis and senescence in type II AECs contribute to the emphysema observed in patients with COPD. Questions remain about how autophagy alters the contributions of epithelial, mesenchymal and other cell populations to lung regeneration during the ageing process.
Autophagy maintains genomic stability
The accumulation of genetic damage throughout life is a common denominator of ageing and genomic instability is a primary hallmark of ageing [8]. DNA damage may affect essential genes and transcriptional pathways, cause subevent dysfunction in cells and, if not eliminated, may jeopardise homeostasis in cells.
DNA damage is prominent in the lung cells of patients with COPD under a hypoxic response condition [89]. Increased breaks in DNA double strands and DNA repair inefficiency may be associated with increased levels of oxidative stress; these are correlated with an increased susceptibility to the development and progression of diseases [90]. Furthermore, mitochondrial DNA (mtDNA) damage and apoptosis of AECs mediated by Sirtuin (SIRT) 3 deficiency promote PF [91]. Genome instability also amplified by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. The coronavirus papain-like protease degrades transcription factor p53 and allows the replication of infected cells [92], while, in addition to participating in the DNA damage response (DDR) and contributing to the ageing process [93], p53 can downregulate coronavirus replication by regulating the cell cycle [94].
The DDR is an evolutionary conserved signalling network responsible for DNA lesion sensing and DNA repair. DDR-induced autophagy selectively degrades distinct proteins, such as ribonucleotide reductase, ubiquitin-specific protease 14 and checkpoint kinase 1, which directly or indirectly influence DNA repair and cell fate, such as proliferation, apoptosis and senescence [95–97]. DNA can be directly degraded through autophagy. There also exists nucleophagy, which is conserved in mammalian cells [98] and may be a defence mechanism that protects cells from tumorigenesis.
Autophagy has a protective effect against DNA damage in ageing-related lung diseases. For example, CS is a strong risk factor for IPF and is also a pro-senescent factor. Zhang et al. [99] found that CSE reduced autophagy and mitophagy and increased mitochondrial ROS in murine lung epithelial-12 cells, and that CSE promoted DNA damage, downregulated tNAD+/NADH ratio and suppressed SIRT1 activity. Autophagy inducer rapamycin and the mitochondria-targeted antioxidant mitoquinone inhibited CSE-related senescence and decreased mitochondrial ROS and that activating SIRT1 attenuated senescence through an autophagy-dependent pathway [99]. In sepsis-induced ALI, the level of circulating mtDNA and the degree of STING (stimulator of interferon genes) activation are increased, mtDNA evoked an inflammatory storm and disturbed autophagy, inducing autophagy or STING deficiency can markedly alleviate lung injury [100]. These data suggest that autophagic modulation of DNA haemostasis is essential for health improvement, ageing and the prevention of diseases in the lungs.
Mitophagy regulates mitochondrial quality control
Mitochondria are “powerhouses” that produce cellular energy in the form of ATP. As cells and organisms age, the efficacy of the electron transport chain (ETC) tends to reduce, accompanied by increased electron leakage and reduced ATP generation [101]. Mitochondrial dysfunction contributes to physical ageing and a wide spectrum of age-related diseases, particularly lung diseases [102]. Mitochondrial dysfunction may increase the risk of severe COVID-19 outcomes; in those who have COVID-19, the increased energy expenditure secondary to a cytokine storm can lead to a nonadaptive state, overwhelming the metabolic reserve capacity of the mitochondria [103].
One study showed that the activation of mitophagy is required to extend the lifespan in several long-lived C. elegans mutants, including the insulin-like growth factor (IGF)/IGF-1 mutants [104]. In the lungs, mitophagy plays an important role in the pathogenesis of ageing-associated pulmonary disorders. The knockdown of both PINK1 and PARK2 enhances the senescence of human bronchial epithelial cells (HBECs) in response to CSE and is accompanied by accumulated damage to the mitochondria and increased ROS production [44]. Moreover, impaired mitophagy also participates in the regulation of myofibroblast differentiation in lung fibroblasts [87, 105]. Insufficient mitophagy due to PARK2 deficiency induces mtROS production, accompanied by activated platelet-derived growth factor receptor/mammalian target of rapamycin (mTOR) signalling, which causes myofibroblast differentiation and proliferation [105].
Autophagy regulates cellular senescence
Increased cellular senescence is a hallmark of ageing [8]. As the number of senescent cells increases with age, senescence may be a beneficial compensatory response to rid tissues of damaged and potentially oncogenic cells. However, this cellular checkpoint also requires an efficient cell replacement system that contributes to the clearance of senescent cells and mobilisation of progenitors to re-establish cell numbers [8]. Senescent cells are apoptosis-resistant and secrete numerous factors termed the “senescence-associated secretory phenotype” (SASP) [106, 107]. The SASP mediates many of their pathophysiological effects and contributes to age-related conditions [108].
In the lungs, mouse studies showed the causal relation of senescence to lung ageing and diseases; senescent-prone recombinant mice exhibit accelerated lung ageing and are more susceptible to experimental lung injury [109, 110]. In contrast, the ablation of senescence cells in the lung in vivo prolongs life and has a restorative effect on lung tissue [111, 112]. In IPF, lung fibrosis is mediated, in part, by senescent cells; cellular senescence markers are detectable within lung tissue and senescent cell deletion rejuvenates pulmonary health in old mice [69]. Furthermore, cellular senescence has been implicated in the pathogenesis of COPD and asthma [113, 114]. Recently, Lee et al. [115] found that virus-induced senescence is also a pathogenic trigger of COVID-19-related cytokine escalation and organ damage, and senolytic targeting of virus-infected cells can be a treatment option against SARS-CoV-2 and other viral infections.
The detailed molecular mechanism of the regulation of cellular senescence is complex and yet to be clarified; it has been proven that autophagy insufficiency plays a pivotal role in the accumulation of deleterious cellular components—as one of the typical manifestations of cellular senescence is the accumulation of damaged proteins and organelles, occasionally associated with ubiquitinated aggregations. Cellular senescence induces autophagy through the inhibition of mTOR and the activation of AMPK and SIRT1 [116, 117]. SIRT1 deacetylation of Atg proteins and transcription factors is involved in autophagy induction [118].
In lung diseases, such as CS-induced COPD, CSE transiently induces autophagy activation with subsequent accumulation of sequestosome-1 and ubiquitinated proteins and an increase in HBEC senescence. The inhibition of autophagy further enhances HBEC senescence, accompanied by the accumulation of sequestosome-1 and ubiquitinated proteins, which reflect insufficient autophagic degradation; moreover, the knockdown of both PINK1 and PARK2 enhances HBEC senescence in response to CSE, accompanied by the accumulation of damaged mitochondria and increased ROS production [44]. However, other studies showed that autophagy may facilitate cellular senescence and that autophagy and senescence may occur in parallel. For example, the overexpression of the Atg gene Unc-51 like autophagy activating kinase (ULK) 3 can induce both autophagy and senescence, whereas the inhibition of autophagy delayed the senescence phenotype, including the SASP [119, 120]. Nonetheless, it can be inferred that autophagy and cellular senescence are interrelated and these two important cellular processes may be dependently and interdependently involved in the pathophysiology of lung diseases.
Autophagy regulates inflammation and inflammageing
Inflammation is an adaptation mechanism designed to maintain organismal homeostasis and inhibit acute and local perturbations, specifically in response to infection or injury [121]. “Inflammageing”, first termed by Franceschi et al. [122], describes a chronic, sterile, low-grade inflammation that contributes to the pathogenesis of age-related diseases. In the lungs, inflammation arises due to persistent exposure to various stimuli and stress. For example, the “cytokine storm” frequently observed in patients with ARDS with severe COVID-19 infection is thought to be a consequence of inflammageing [123, 124]. Older individuals are more susceptible to lung injury due to dysregulated response compared with that in younger individuals.
The exact origin and drivers of inflammageing remain yet to be clarified and may involve multiple ageing mechanisms that occur throughout the body (figure 3). Franceschi et al. [122] proposed that in the ageing process, the accumulation of “cellular garbage”, represented by endogenous/self, misplaced or altered molecules due to damaged and/or dead cells and organelles (cell debris), acts as damage-associated molecular patterns (DAMPs) that activate an auto-inflammatory response by binding to pattern recognition receptors (PRRs) on innate immune cells, particularly macrophages [122]. Age-related declines in autophagy and macrophage phagocytic activity contribute to DAMP accumulation and lead to inflammation and ageing.
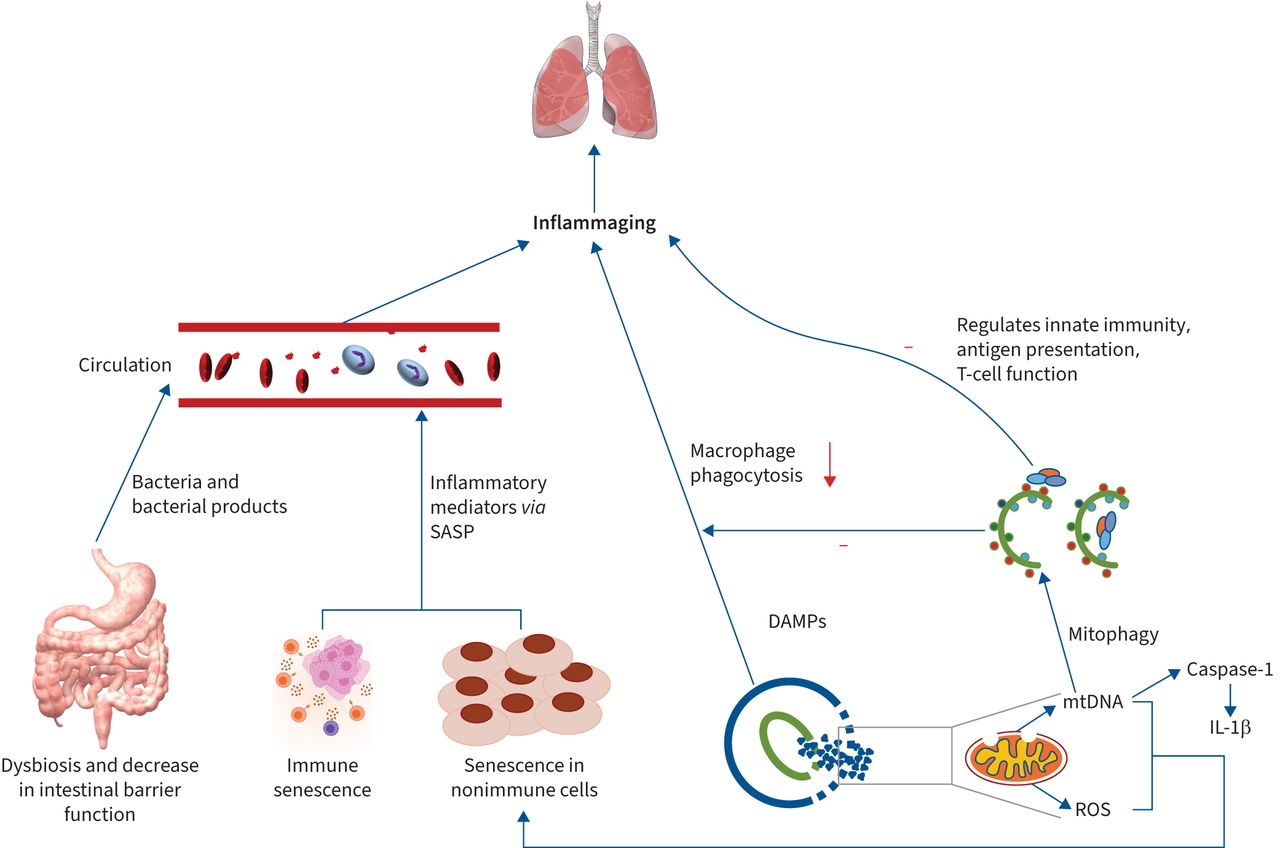
Multiple mechanisms contributing to inflammaging and the involvement of autophagy. Multiple mechanisms contribute to inflammaging, eventually leading to lung pathogenesis. Senescence in immune and nonimmune cells increases inflammatory mediator release via the release of senescence-associated secretory phenotype (SASP). In aged intestines, dysbiosis of intestinal microbiota and a decreased intestinal barrier allow bacterial products to translocate to the circulatory system, where they trigger low systemic inflammation. Damaged and/or dead cells and organelles act as damage-associated molecular patterns (DAMPs) to activate an auto-inflammatory response by binding to pattern recognition receptors (PRRs) on innate immune cells. Age-related declines in autophagy and macrophage phagocytic activity also contribute to DAMP accumulation and lead to inflammation and ageing. Autophagy promotes recycling cellular content, generating nutrients and energy to maintain homeostasis, preventing recognition of pathogen-associated molecular patterns by PRRs and inflammation. Autophagy can also control inflammation via the regulation of innate immunity signalling by removing endogenous inflammasome agonists and through its effects on immune mediator secretion. Additionally, autophagy modulates innate immunity, antigen presentation and T-cell function. Furthermore, mitophagy inhibits mitochondrial DNA (mtDNA) accumulation and excessive mitochondrial reactive oxygen species (ROS) production, both of which may accelerate cellular senescence and aggravate the inflammaging process. IL-1β: interleukin 1 beta.
By eliminating the debris and products of cellular metabolism, autophagy promotes the recycling of cellular content, thus generating nutrients and energy to maintain homeostasis and preventing the recognition of DAMPs by PRRs and the consequent inflammation [125]. Furthermore, autophagy could control inflammation through the regulation of innate immunity signalling by removing endogenous inflammasome agonists and through effects on the section of immune mediators. Additionally, autophagy modulates antigen presentation and T-cell homeostasis and affects T-cell repertories and polarisation [126]. The promotion of autophagy through starvation or rapamycin administration inhibits the activation of inflammasome [127].
Autophagy regulates oxidative stress
In biological systems, oxidative stress is generated when endogenous antioxidant defences are impaired and/or overwhelmed by the presence of ROS [128]. Ageing hallmarks contributing to the ageing process could also be caused by oxidative stress. For example, telomeres are highly sensitive to oxidative stress and their repair capacity is poor compared with that of other parts of the chromosome [129].
Autophagy is activated in response to oxidative stress to protect the cells from apoptosis [130]. Increased oxidative stress is sensed by the lysosomal cation channel, mucolipin 1 [131]. Mucolipin 1 oxidation promotes channel opening; Ca2+ is released from the lysosomal lumen and the Ca2+-dependent phosphatase, calcineurin, is activated [132]. Calcineurin-dependent transcription factor E (TFEB) phosphorylation promotes TFEB translocation to the nucleus and stimulates autophagy [132]. ROS can also activate autophagy by inhibiting the PI3K-Akt or by activating AMPK to inhibit the mTOR signalling pathway [133]. Lung diseases are inevitably accompanied by hypoxia, which is a major stimulus for the induction of autophagy [134]. Hypoxia can impair mitochondrial ETC activity, increase mitochondrial O2⋅− production and result in mitochondrial dysfunction [134, 135]. Chronic and moderate hypoxia trigger hypoxia-inducible factor-1α and delta isoform of protein kinase C–JNK1-mediated pathways to activate autophagy [136]. Oxidative stress has been identified as the major contributor to the dysregulated response and intractable inflammation under CSE [137]. CSE causes mitochondrial damage and accumulation by impairing mitophagy in a deteriorative state; mitophagy protects HBECs by removing damaged mitochondria and reducing the production of ROS [44].
Collectively, autophagy represents a negative feedback mechanism of regulation, which eliminates the accumulation of ROS and protects cells from oxidative injury; this is particularly critical in ageing-related lung diseases.
Autophagy regulates immune function
The immune system cannot escape from the effects of ageing and displays senescence characteristics in aged individuals, that is, immunosenescence, producing a chronic inflammatory status of the organism, as manifested by a decreased ability to counteract antigens [138].
Immunosenescence and a corresponding decrease in immune regulation are implicated in age-related lung diseases. Elderly individuals face a greater risk of many diseases, particularly respiratory diseases, due to their poor response to immune challenges compared with younger ones.
Autophagic activity decreases with age in immune cells. Decreased activities of autophagy and mitophagy in phagocytic cells may impair their ability to kill pathogens and arouse dysregulated activation of inflammasomes, which increases the production of inflammatory cytokines and contributes to the age-related inflammageing phenotype.
The human respiratory tract is an important immune interface requiring a tightly regulated response to the continuous exposure to environmental stress [2]. Manoeuvring autophagy to tune immune function and immunosenescence can be a promising therapeutic management in ageing and ageing-related lung diseases.
Conclusions
Autophagy plays a central role in the regulation of ageing. Pulmonary diseases are characterised by a dysregulation of autophagic activity, particularly in those at an advanced age in several species. The detailed mechanism of autophagy in physiological ageing and pathological conditions still need to be further explored.
Enhancing autophagy generally promotes cellular functions and homeostasis, thereby prolonging lifespan and improving pulmonary health. However, it should be noted that significantly increasing autophagy may shorten the lifespan and harm the lungs [23]. Targeting autophagy for therapeutic exploitation in ageing and ageing-related lung diseases depends on the exact nature of the autophagic defect present in each cell type. In the studies discussed in this review, the exact autophagic defects contributing to ageing-related lung diseases are yet to be fully elucidated. This can be partly attributed to the limited specificity of current autophagy modulators in targeting one cell type in lung tissue.
Based on current literature, it can be hypothesised that the possibility of improving autophagy activity to boost cell function in lungs offers a very attractive therapeutic approach to enhance lung function in the elderly. For example, modulating autophagy through calorie restriction, exercise can be a promising therapeutic agent for various pulmonary diseases [139, 140]. Long-term health benefits for the lungs will likely arise from achieving the right balance of autophagy, which will depend on lung tissue and organismal age. Furthermore, the use of autophagy modulators (inductors/inhibitors) combined with other diverse therapies provides a promising strategy to treat a variety of pulmonary conditions.
Footnotes
Provenance: Submitted article, peer reviewed.
Published in volume 31, issue 166 of the European Respiratory Review on 21 December 2022; republished 5 January 2023 with amendments to the authors' affiliation details.
Author contributions: Z. Fu formulated the idea and collected the literature, Y. Zhang organised the paper and created the figures, and Z. Fu wrote the manuscript. J. Zhang reviewed the manuscript. All authors read and approved the final manuscript.
Conflicts of interest: The authors declare no conflict of interest.
- Received July 20, 2022.
- Accepted November 8, 2022.
- Copyright ©The authors 2022
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org











{kind=link}
{kind=link}
{kind=link}