





Abstract
There is a need for clinical trial end-points to better assess how patients feel and function, so that interventions can be developed which alleviate symptoms and improve quality of life. Use of 6-min walk test (6MWT) outcomes as a primary end-point in interstitial lung disease (ILD) trials is growing, particularly for drugs targeting concurrent pulmonary hypertension. However, 6MWT outcomes may be influenced differentially by interstitial lung and pulmonary vascular components of ILD, making interpretation complicated. We propose that using 6MWT outcomes, including 6-min walk distance or oxygen desaturation, as primary end-points should depend upon the study population (how advanced the ILD is; whether vasculopathy is significant), the degree of disease progression, and, importantly, the effect of study treatment expected. We argue that the 6MWT as a single outcome measure is suitable as a primary end-point if the treatment goal is to improve functional performance or prevent disease progression within a study population of patients with advanced ILD or those with ILD and co-existent vasculopathy. In addition, we discuss the potential of composite primary end-points incorporating 6MWT outcomes, outlining important considerations to ensure that they are appropriate for the study population and treatment goals.
Abstract
The use of the 6-min walk test as a primary end-point in interstitial lung disease clinical trials should depend upon the study population, the degree of disease progression expected and how this might be impacted by the study treatment https://bit.ly/39Dp1ig
Introduction
The 6-min walk test (6MWT) is used to objectively measure functional exercise capacity, assess prognosis and evaluate treatment response [1, 2]. Historically, the 6MWT has been widely used in clinical trials of pulmonary arterial hypertension (PAH) populations. Although there are other measures of exercise or functional capacity, such as shuttle walk tests and cardiopulmonary exercise tests [3], the 6MWT is the measure with which most experience has been gained within a clinical trial setting [4, 5]. The 6MWT is becoming increasingly used in chronic respiratory disease settings, including COPD and interstitial lung diseases (ILDs) [2].
ILDs are characterised by fibrosis and/or inflammation of lung tissue, and have heterogeneous interstitial lung and pulmonary vascular components that contribute to clinical disease manifestations [6]. Patients with ILD often develop concurrent pulmonary hypertension (PH), which is associated with an increased need for supplemental oxygen, reduced mobility and decreased survival [7]. The development of mild ILD manifestations in patients with PAH has been reported to have a negative impact on survival and treatment response [8].
Most ILD trials in recent years have used change in lung function measured using forced vital capacity (FVC) as the primary end-point, which is regarded by regulators as an acceptable surrogate end-point for clinical outcomes [9–11]. However, FVC only captures one domain of disease progression and does not mirror change in quality of life (QoL). Patients do not directly perceive improvement or decline in physiological measurements such as FVC. As such, other end-points are needed that better assess how the patient feels and functions in order to develop interventions that provide benefit in terms of symptoms and QoL [9, 12]. A 2015 report from the United States Food and Drug Administration emphasised the need for such end-points as part of the patient-focused drug development initiative, summarising that future clinical trial outcome measures should focus on improving symptoms, QoL and patients’ day-to-day functioning [9, 12]. While approved therapies have successfully slowed FVC decline, they have not been shown to impact patient function [9]. An improvement in functional capacity, as measured by the 6MWT, has demonstrated a clear connection with health improvement in patients with idiopathic pulmonary fibrosis (IPF) [9], and has shown a significant correlation with health-related QoL (p<0.001) [13].
The use of 6MWT outcomes as a primary end-point in ILD trials is growing, but this has historically been restricted mainly to PAH drug trials with the aim of targeting any concurrent PH [1, 14–20]. However, using the 6MWT in ILD is more complex than in PAH, and findings in PAH cannot be extrapolated to ILD. Crucially, 6MWT outcomes may be influenced differentially by interstitial lung and pulmonary vascular components of ILD [6]. Thus, 6MWT outcomes may mean different things in ILD without vasculopathy, ILD with vasculopathy and pure vasculopathy. Therefore, the 6MWT is a valuable “catch-all” variable that, in our opinion, and with recognition that it is not the most appropriate primary end-point in some patient populations, should be included as a secondary end-point in all ILD trials.
Here, we address the use of the 6MWT as a primary end-point in ILD drug trials, proposing that the choice of end-point should depend upon the study population (how advanced the ILD is; whether vasculopathy is significant), the degree of disease progression expected during the study, and how this might be affected by study treatment. We argue that the 6MWT as a single outcome measure (e.g. 6-min walk distance (6MWD) or oxygen desaturation) is suitable as a primary end-point if the treatment goal is to improve clinical performance or prevent rapid disease progression in the study population. We advocate the use of the 6MWT as a primary end-point in patients with advanced ILD and those with coexistent vasculopathy (ILD-PH; including sarcoidosis) as a global measure of progression in advanced disease, when further meaningful decrements in lung function are limited by a “floor effect” (a lower limit to the data, below which values cannot be reliably specified) in the FVC. In study populations that are likely to have a short 6MWD, we speculate that oxygen desaturation during the 6MWT may be valuable in reliably detecting change, although further research is needed to confirm the potential of this as a primary end-point in ILD populations. In addition, we discuss the potential of composite end-points incorporating 6MWT outcomes, outlining the two types of composites and how including both types as co-primary end-points could ensure that they are clinically relevant, robust and appropriate across a range of ILD populations and treatment goals. This review focuses only on fibrosing ILD populations, since limited evidence is available to discuss the role of the 6MWT in nonfibrosing ILDs.
Search strategy and selection criteria
Relevant references for this review were identified through PubMed searches during August 2021 using the terms “six-minute walk test”, “six-minute walk distance”, “6MWT”, “6MWD” or similar variations. Deeper searches included the terms: “idiopathic pulmonary fibrosis”, “interstitial” or similar variations; “multivariate” or “independent”; “pulmonary arterial hypertension”, “PAH” or “pulmonary hypertension”; and “randomised”, “randomized” or the Clinical Trial filter. No limits were placed on date or language.
Conduct and use of the 6MWT in clinical practice
The 6MWT involves a patient walking as far as possible (without exhaustion) over 6 min on a hard, flat surface, such as a hospital corridor (figure 1) [2, 22]. The 6MWD is recorded in metres; other 6MWT outcomes that are typically recorded include peripheral oxygen saturation (SpO2), heart rate and dyspnoea (measured using the Borg scale) [2].

The 6MWT is simple and inexpensive to perform, reproducible, replicates the activities of daily living and provides an objective measure of the patient's overall cardiopulmonary reserve [22]. While results do not provide insight into the underlying reason for exercise limitation, the 6MWT is valuable as a snapshot of the individual's functional exercise capacity and as a means to measure changes over time [22]. However, 6MWT results can be affected by multiple factors including methodological variations (e.g. indoors versus outdoors, track length); patient demographics (age, height, sex, weight); comorbidities that affect walking ability or oxygen desaturation; the use of supplemental oxygen; language barriers; and training effects [2, 22]. These limitations are further discussed in the European Respiratory Society and the American Thoracic Society technical standard [2].
The 6MWT has not been fully standardised as there is no consensus for certain aspects, including how to manage supplementary oxygen use during the test and whether the 6MWT should be stopped if SpO2 is low [23, 24]. Since the 6MWT is highly sensitive to changes in methodology [2, 22, 24], the resulting variability of 6MWD measurements may lead to a loss of precision. This may be problematic in patients with advanced ILD (with or without vasculopathy), since baseline 6MWD may be too short to yield a clinically relevant and reliable change over time [25, 26]. Thus, standardisation of the 6MWT and its consistent implementation within a trial is particularly important when using 6MWT outcomes as a primary end-point. An approach for standardisation of the 6MWT in IPF trials has been proposed, and was under evaluation in the ISABELA trials prior to their termination [24]. This standardised approach suggested that supplementary oxygen flow rate should be based on a titration procedure and kept consistent throughout the study, and the 6MWT should only be stopped when SpO2 falls below 80% for 15 s or if the patient experiences certain symptoms [24].
Predictive factors for 6MWT outcomes and their prognostic value
In ILD, real-world multivariate analyses have identified a variety of disease attributes that correlate with 6MWT outcomes, including diffusing capacity of the lung for carbon monoxide (DLCO), mean pulmonary artery pressure and right ventricular systolic pressure (table 1) [27–33]. Further analyses have determined that 6MWT outcomes have prognostic value in patients with ILD from real-world or clinical trial populations (table 2) [13, 30, 33–44]. A substantial body of evidence demonstrates that baseline 6MWD is independently associated with higher mortality in patients with IPF, with the cut-off for an association ranging from ∼200 m to 300 m [34–37, 43, 44]. However, other studies have not shown this association [13, 30, 33, 38–41]. SpO2 during the test, an impaired chronotropic response and worse heart-rate recovery after 1 min have all been shown to have prognostic value for mortality [30, 33, 36, 38–41].
Predictive factors for 6-min walk test (6MWT) outcomes in patients with interstitial lung disease (ILD) in real-world studies
Prognostic value of 6-min walk test (6MWT) outcomes in patients with interstitial lung disease (ILD) from real-world studies and clinical trials
These findings support the clinical relevance of including oxygen desaturation and heart rate in addition to 6MWD in the 6MWT operating procedures for patients with ILD. The findings that DLCO and haemodynamic parameters were predictive for certain 6MWT outcomes, and that these outcomes were prognostic for mortality, are to be expected from the underlying interstitial lung and pulmonary vascular components seen in patients with ILD [6].
What 6MWT outcomes and DLCO tell us about ILD progression
Both the 6MWT and DLCO encompass interstitial lung and pulmonary vascular components of ILD, unlike FVC, which seems to be more specific to the interstitial lung component [6, 45]. It is possible to have mild ILD with severe vasculopathy or advanced ILD without vasculopathy, and it might not be possible to distinguish between these using 6MWD or DLCO alone. In a retrospective study in patients with IPF, the 6MWT alone could not distinguish between patients with or without PH as there was no reported difference in 6MWD between these two groups [25]. However, a significant linear correlation was found between DLCO and 6MWD, which the authors hypothesised was due to variations in DLCO that may be related to both parenchymal lung involvement and vascular changes [25]. The authors summarised that little is known of the influence of PH on 6MWT performance, especially as to whether it may reduce exercise capacity in patients with IPF or reflect the presence of advanced lung disease [25]. Conversely, the 6MWT has demonstrated a difference between patients with ILD versus COPD, based on arterial desaturation during the 6MWT [46]. However, the degree of exercise-induced arterial hypoxaemia varies between diseases and ILD subtypes [47–49]. Since 6MWD can vary widely, regardless of lung-function impairment severity measured by FVC [45], different phenotypes of ILD may exist depending on the balance between interstitial lung and pulmonary vascular components. With 6MWD, there is no indication as to which of these components is driving changes. Therefore, confusion could arise when using 6MWD as an end-point in a trial where there is a mix in the balance of these two components of ILD in the population, particularly when the study treatment does not lead to a clear improvement in 6MWD.
For assessment of individual patients in clinical practice, interpretation of 6MWT data should include integrating a wide range of measurements to determine what an abnormal result may mean. To risk stratify patients for underlying PH, physicians can compare 6MWD with oxygen desaturation and account for carbon monoxide transfer coefficient or FVC/DLCO ratio alongside assessing other parameters, including markers of pulmonary vasculopathy (e.g. alveolar–arterial gradient for oxygen (PA–aO2) at rest, brain natriuretic peptide levels, pulmonary artery dilation on computed tomography, echocardiography findings); markers of ILD severity; and presence of systemic comorbidities, including those linked to the primary disease (e.g. connective tissue disease (CTD)).
Use of the 6MWT as a primary end-point in clinical trials
PAH trials
Historically, 6MWD was the most common primary end-point in PAH trials, which has led to the approval of many PAH drugs [5, 50–63]. However, change in 6MWD had limitations as a primary end-point in some trials, where the change was less than the now recognised minimal clinically meaningful difference (MCID) [5, 64] despite any statistical significance achieved [53–58, 60, 65–67]. Typically, PAH studies have not included oxygen desaturation as part of the primary end-point or systematically accounted for supplemental oxygen use during the 6MWT.
More recent phase 3 trials, such as those leading to approval of newer PAH drugs, included categorical 6MWD decline as part of a composite primary end-point [65–68], which incorporated mortality and morbidity outcomes (e.g. worsening functional class, hospitalisation, initiation of prostanoid therapy, lung transplantation/atrial septostomy) and 6MWD decline [5, 65–68]. Composites reflecting time to clinical worsening have now been widely accepted as primary end-points for PAH drug registration trials [5], although 6MWD alone is still used in early proof-of-concept studies. However, the definition of time to clinical worsening has not been standardised across trials, individual components have not been weighted against their clinical importance and frequency of occurrence, and the relationship between clinical worsening and subsequent mortality is not well-defined [5].
ILD trials
Unlike in PAH trials, the 6MWT has not been the gold-standard outcome for IPF/ILD trials and using a 6MWT outcome as the only primary end-point in ILD trials is more complicated. Interventions that have been assessed in ILD have generally been specific to either the interstitial lung (antifibrotics) or pulmonary vascular component (PAH drugs) of ILD.
To date, ILD trials using the 6MWT as a primary end-point have mostly evaluated PAH drugs (except for two phase 2 trials assessing pirfenidone in patients with IPF [14, 18]). These drugs were either chosen to target PH in patients with advanced IPF or ILD-PH (STEP-IPF, RISE-IIP, INCREASE) [17, 19, 20], or to assess a possible antifibrotic action of the endothelin receptor antagonist, bosentan, in more general ILD populations excluding advanced disease (BUILD-1, BUILD-2; table 3) [15, 16]. ILD trials using the 6MWT as a primary end-point differed in their 6MWT methodology and/or did not publish much information on 6MWT methodology, making comparisons between trials challenging [14–20, 24]. Since trials largely evaluated PAH drugs, findings cannot be extrapolated to antifibrotics or other drugs not primarily addressing the pulmonary vasculature.
Interstitial lung disease (ILD) randomised controlled trials using 6-min walk test (6MWT) as the primary end-point
Until recently, primary end-points based on 6MWT outcomes were not met in ILD trials (table 3) [14–19]. A subgroup analysis of STEP-IPF assessing patients with echocardiograms available for independent review found that 6MWD decline was significantly less with sildenafil versus placebo in the subgroup with right ventricular systolic dysfunction, but not the subgroup with right ventricular hypertrophy [69]. Recently, the primary end-point of change in 6MWD over 16 weeks was met in the INCREASE study, which investigated inhaled treprostinil in patients with ILD-PH [20], indicating that the 6MWT may have a place as a primary end-point in some ILD populations, specifically those with advanced ILD or ILD-PH.
In other ILD populations aside from IPF, there may be specific limitations of the 6MWT. In CTD-ILD the presence of various extrapulmonary features (skin fibrosis, musculoskeletal pain, heart involvement, anaemia) and associated disability may confound interpretation of the 6MWT [70]. Nevertheless, in the INCREASE study, patients (n=326) with various lung diseases, including CTD (n=72) and concurrent PH, demonstrated significant improvements in 6MWD following treatment with inhaled treprostinil [20]. Treatment targeting concurrent PH improved 6MWD; therefore, it is likely that change in 6MWD reflects improved pulmonary haemodynamics with exercise. In sarcoidosis-associated PH, the 6MWT captured the global effects of sarcoidosis, with prognostic value that correlated with other physiological and haemodynamic variables [71]. However, further evidence on the relevance of the 6MWT in other ILD populations is limited and remains an unmet need.
Proposed place of the 6MWT as a primary end-point in ILD trials
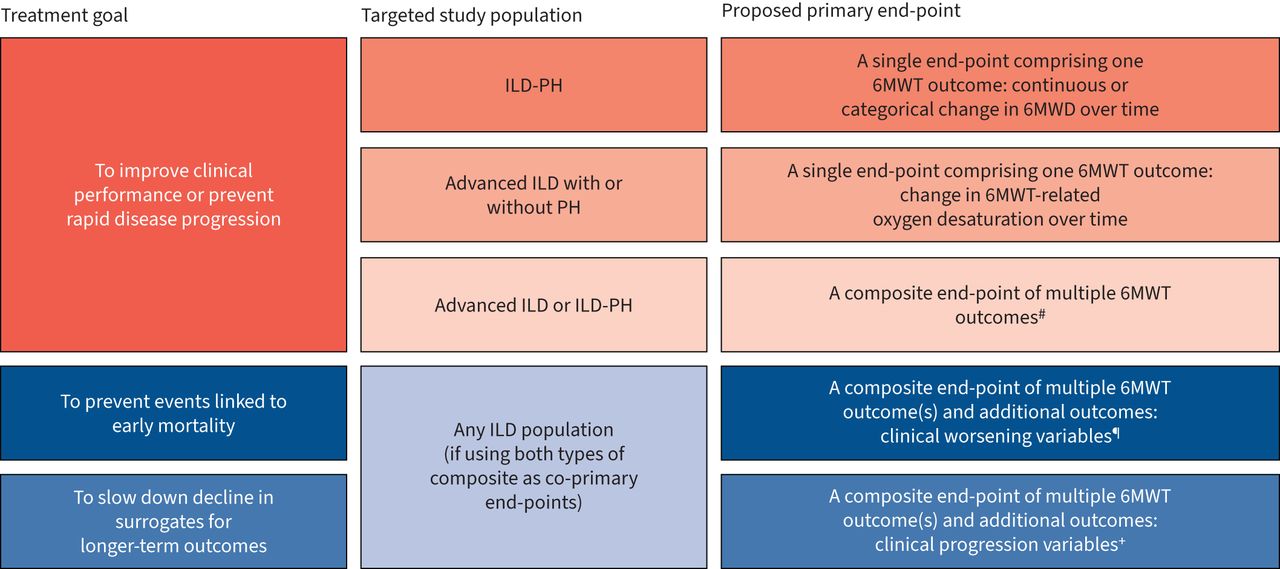
We propose that the place of the 6MWT as a primary end-point in ILD trials requires consideration of the study population (i.e. how advanced the ILD is; whether vasculopathy is significant), the degree of disease progression expected during the study and how this might be affected by the study treatment (figure 2).
{kind=link}
{kind=link}
Potential uses of the 6-min walk test (6MWT) as a primary end-point in interstitial lung disease (ILD) trials. PH: pulmonary hypertension; 6MWD: 6-min walk distance. #: e.g. distance–saturation product; categorical change in 6MWD accompanied by a decrease in the peripheral oxygen saturation or increase in the Borg score. ¶: e.g. death, respiratory-related nonelective hospitalisation, acute exacerbations, large categorical declines in forced vital capacity (FVC) and/or 6MWT (6MWD, oxygen desaturation or a combination of 6MWT outcomes). +: e.g. categorical declines in FVC, diffusing capacity of the lung for carbon monoxide and/or 6MWT (6MWD, oxygen desaturation or a combination of 6MWT outcomes) that are smaller than those associated with clinical worsening.
6MWD as a primary end-point
Categorical change in 6MWD over time may have potential as a primary end-point in trials assessing a treatment that is expected to improve clinical performance or prevent rapid disease progression in the study population (particularly patients with advanced ILD or ILD-PH).
An assumption we challenge is that change in 6MWD can be used and interpreted universally across fibrotic ILD trials, regardless of the condition, disease severity and expected effect of study treatment on disease progression. 6MWD offers the benefit of representing a “catch-all” that mirrors everyday exercise tolerance and how the individual functions. INSPIRE and CAPACITY found that 6MWD correlated with dyspnoea (by University of California, San Diego Shortness of Breath Questionnaire) and QoL (by St George's Respiratory Questionnaire), as well as FVC, DLCO and resting PA–aO2 [13, 43]. Nonetheless, a primary end-point should be specific to the intended direct action of a therapy, unless there is a compelling argument otherwise. In PAH, 6MWD is specific to changes in vasculopathy, whereas in ILD, 6MWD reflects changes in both ILD severity and vasculopathy [6, 20, 72].
The INCREASE study showed that 6MWD may be a suitable primary end-point in trials using a PAH drug to target vasculopathy in patients with ILD-PH [20]. In INCREASE, 6MWD increased by 21.1 m with inhaled treprostinil over the 16-week treatment period, but decreased by 10.0 m with placebo; thus, not only did the treatment prevent disease progression, it also improved functional ability [20]. A slight increase in FVC was also observed with inhaled treprostinil over the 16-week treatment period, particularly in the subgroup of patients with idiopathic interstitial pneumonia (IIP), compared with a decrease in FVC with placebo. This may be due to decreased intravascular pressure and/or pulmonary vascular resistance with inhaled treprostinil treatment, perhaps suggesting that pulmonary vasculopathy may have a minor effect on FVC [73]. Alternatively, endothelial pathways may contribute to the progression of interstitial fibrosis, or treprostinil may itself have antifibrotic properties as shown in two animal studies where treprostinil inhibited fibroblast activity and attenuated vascular remodelling and fibrosis [74–76]. A recent study in patients with fibrotic ILD at risk of PH also found that patients who received pulsed inhaled nitric oxide had improved physical activity (assessed by the level of moderate/vigorous physical activity) over 8 weeks, whereas physical activity level with placebo decreased [77]. Improvements in 6MWD and SpO2 were observed with inhaled nitric oxide versus placebo, but these differences were not statistically significant [77]. Therefore, we believe that 6MWD may be best placed as a primary end-point when the study drug is expected to improve clinical performance, as well as to possibly prevent disease progression. This makes the 6MWT as a primary end-point particularly relevant in patient populations with advanced ILD or ILD-PH.
Since increased categorical 6MWD decline has been shown with greater consistency to predict mortality in ILD than baseline 6MWD [13, 30, 33–41, 43, 44], using categorical 6MWD decline may be preferable to mean 6MWD decline in a primary end-point. The reason for which could be that a large 6MWD decline is needed to confirm the change is directly linked to ILD progression rather than the influence of other factors on 6MWD. For the MCID of the 6MWD in IPF trials, distribution- and anchor-based analysis of 6MWD has yielded estimates ranging between 21.7 and 37 m, and a value of 28 m yielded from the mean of all MCID point estimates [13, 78]. Further evidence is required to establish the MCID in 6MWD in non-IPF ILD. Findings from the INSPIRE study in an IPF population (with baseline FVC ≥55% and 6MWD ≥150 m) support that change in 6MWD over time is predictive for mortality [43, 44]. The MCID for 6MWD decline was estimated between 24 and 45 m based on the predictive value of change in 6MWD over 48 weeks for hospitalisation and/or mortality [43]. A categorical 6MWD decline of >50 m was associated with greater 1-year mortality [43, 44]. Similar findings were observed in the placebo arms of CAPACITY in patients with IPF, again all of whom had baseline 6MWD ≥150 m [13]. However, a categorical 6MWD decline of >50 m might be too large to determine a treatment effect for patients with advanced disease (who are likely to have a low baseline 6MWD). To overcome this issue, a relative categorical decline could be implemented, such as a relative decline of ≥15%, which was used as part of the composite end-point in a trial of sildenafil added to pirfenidone in patients with advanced IPF and risk of PH [1]. We propose that a relative categorical 6MWD decline might be better for populations that include patients with advanced IPF.
Categorical end-points have limitations, including lack of consensus on what constitutes a clinically meaningful change in 6MWD, demonstrated by the range of estimates for MCID. There are no further data to support the use of a relative categorical 6MWD decline, although it has been used routinely as a primary end-point in clinical trials of PAH, where a threshold of 10–20% has been widely used to designate worsening [5, 51, 53, 56–58, 61]. Furthermore, a relative decrease in 6MWD >15% from baseline was used as a composite end-point for time to clinical worsening in the INCREASE study [20]. Recently, it has been suggested to define advanced ILD based on 6MWD criteria and oxygen needs during the 6MWT rather than FVC, since one study found that functional impairment (measured by 6MWD) varied widely regardless of physiological impairment (measured by FVC) [45]. Likewise, to avoid inclusion of patients with baseline 6MWD that is too short to observe a reliable change over time, we recommend restricting recruitment based on 6MWD criteria and oxygen needs during the 6MWT.
Thresholds are needed for change in the 6MWT when it is included as part of a categorical composite end-point. However, in general, end-points are more sensitive when analysed as continuous variables. For this reason, we favour 6MWT change expressed as a continuous variable when it is a single primary end-point to reduce the likelihood of a false-negative study.
6MWT oxygen desaturation as the primary end-point
Change in oxygen desaturation over time may have potential as an alternative primary end-point to 6MWD in study populations that are likely to have a short 6MWD (advanced ILD with or without PH).
In patients with advanced ILD, 6MWT-related oxygen desaturation has the potential to be a more appropriate measure; greater prognostic value for mortality than 6MWD has been demonstrated in real-world studies [38–41]. In cases where baseline 6MWD is too short to observe a reliable change over time, it could potentially mitigate the problem with measurement variation causing a loss of precision of the 6MWD. Oxygen desaturation could potentially be more relevant than FVC as a primary end-point in an advanced ILD population, since FVC does not reflect changes in functional impairment [45]. Once ILD is advanced, it is possible that further worsening is due to progressive vasculopathy or vessel drop-out; therefore, changes in oxygen desaturation may be better targeted to capture further disease progression than changes in FVC. However, oxygen desaturation has also been found to have poor or modest reproducibility compared with 6MWD in populations with fibrotic IIP or COPD, respectively [40, 79]. Nonetheless, we speculate that oxygen desaturation might be a valuable primary end-point in patients with advanced ILD, with or without PH, since this population is likely to have a shorter 6MWD than those with less advanced ILD.
Further research is needed to confirm the potential of oxygen desaturation during the 6MWT as a primary end-point in ILD populations. This should focus on determining the MCID for change in oxygen desaturation over time, including whether an end-point based on a categorical decline is appropriate, and whether the recovery period in SpO2 (with or without supplemental oxygen) has clinical relevance.
6MWT outcomes as part of a composite primary end-point
Changes in 6MWT-related outcome(s) may have potential as part of clinical worsening and/or clinical progression composites (which we believe should be distinguished); inclusion of both types of composites as co-primary end-points could allow their use in any ILD study population.
To address the limitations of using a single 6MWT-related primary end-point in ILD trials, 6MWT outcome(s) could be part of a composite primary end-point, similar to the evolution of primary end-points in PAH trials [5]. We propose that two types of composite end-points should be distinguished: “clinical worsening” and “clinical progression”. Clinical worsening composites comprise mortality and measures associated with early mortality (increased risk of mortality within the following year, e.g. hospitalisation, major 6MWD and/or FVC change, acute exacerbations), but which may not predict longer-term mortality (increased risk beyond 1 year) well. We propose that clinical progression composites should include measures linked with longer-term mortality (e.g. declines in FVC, DLCO and/or 6MWD that are smaller than those associated with clinical worsening), but which may not predict early mortality well. In oncology, the surrogate end-point progression-free survival (which encompasses mortality and clinical progression) was found to correlate with early mortality in small cell lung cancer and specific advanced cancers but not in other cancers (e.g. nonsmall cell lung cancer) [80, 81], possibly because most patients with less advanced diseases would reach the end-point through clinical progression rather than mortality. Thus, it is important to consider the study population when choosing the type of composite to use as a primary end-point, and to avoid mixing clinical worsening and clinical progression variables in a single end-point.
A composite primary end-point was used in a recent trial (evaluating sildenafil + pirfenidone versus placebo + pirfenidone) in patients with advanced IPF at risk of PH [1]. The primary end-point was disease progression over 52 weeks, defined using a composite of relevant decline from baseline in 6MWD, respiratory-related nonelective hospitalisation or all-cause mortality. Relevant decline from baseline in 6MWD was specified as >25% decline, or 15–25% decline if accompanied during the 6MWT by at least one of the following: worsening of oxygen desaturation or maximum dyspnoea (Borg scale) or increased oxygen requirements [1]. The secondary criteria for relative decline in 6MWD (15–25% decline) was utilised, thereby avoiding a requirement for patients to return to the site to undertake a repeat 6MWT if the decline was close to the threshold for determining a treatment effect [26]. However, addition of sildenafil to pirfenidone provided no treatment difference on the composite primary end-point or its individual components [1].
Composite end-points are widely used in other therapeutic areas, such as cardiovascular disease and oncology [80–82]. However, several problems have been highlighted. Composites often comprise components that differ in clinical importance and frequency, yet the components are often given equal weight (e.g. components of time to clinical worsening end-points in PAH trials) [5, 23, 82]. This means overall treatment effect may be driven by components of lesser clinical importance, since the least serious events would usually occur earlier and with the greatest frequency [82, 83]. Additionally, treatment effects on different components could be directionally different, e.g. a treatment may improve functional end-points yet have adverse effects on mortality, as has been the case for certain cardiovascular treatments (e.g. flosequinan) [82, 83]. The relationship between clinical worsening and subsequent survival may not be well-defined, so composite end-points may not be sufficiently validated as surrogate end-points for mortality [5]. Definitions of composite end-points (e.g. time to clinical worsening) can be variable, which may contribute to heterogeneity in comparing treatment effects across trials [5, 82].
Unlike PAH, the heterogeneity of ILD with interstitial lung and pulmonary vascular components complicates the design of a composite primary end-point and the final result. Determining what type of composite end-point would be more appropriate in a study population may not be possible, as disease progression for ILDs can be variable and hard to predict. One might expect that clinical worsening composites would be particularly suitable for advanced ILD and/or ILD-PH populations, and that clinical progression composites are more suited for less-advanced ILD populations without PH. However, while clinical worsening events are rarer in less-advanced ILD, these events can take place in any patient with ILD, regardless of disease severity, and may not occur in all patients with advanced disease. Thus, it might be most appropriate to have both types of composites as separate co-primary end-points, which could be used for any ILD population since they would encompass both clinical progression and clinical worsening, as well as allow differentiation between clinical worsening that occurs following clinical progression and worsening that occurs without prior evidence of progression. This approach would allow assessment of treatments expected to prevent events linked to early mortality and those expected to slow down decline in surrogates for longer-term outcomes.
Clinical worsening composites could be limited to respiratory-related nonelective hospitalisation and death [45], or could include other clinical worsening variables, such as large categorical declines in 6MWD and/or FVC. Thresholds for categorical decline in 6MWD and FVC over 6 months, which have been associated with greater 1-year mortality, are >50 m and ≥10%, respectively [13, 43, 44, 84]. However, a relative change, such as ≥15%, should be considered for categorical 6MWD decline in advanced ILD populations, as discussed previously. Clinical progression composites could include declines in FVC and/or 6MWT-related end-point(s) over time that are smaller than those associated with clinical worsening, although further research is needed to better define suitable thresholds for these declines. Other variables that could be considered in clinical progression composites include changes in patient-reported outcomes, exertional dyspnoea or findings on computed tomography [23].
Composites of multiple 6MWT outcomes could be considered in trials assessing a treatment that is expected to improve clinical performance or prevent rapid disease progression in the study population, or form part of a wider clinical worsening and/or clinical progression composite. One such 6MWT composite is the distance–saturation product, which is an index that integrates 6MWD and nadir SpO2 during the 6MWT; this has been found to predict mortality more accurately than either component alone [36]. Another potential 6MWT composite is categorical change in 6MWD accompanied by a decrease in SpO2 or an increase in rating on the Borg scale.
For any composite end-point in ILD, we suggest that end-point components could be prospectively weighted with respect to clinical importance and frequency of occurrence [5, 82], e.g. greater weight could be given to hospitalisation or death than to changes in FVC or 6MWT-related outcomes. Individual components of composite end-points should be analysed separately as well as in the composite, thereby ensuring balance between treatment groups in all components and that any harm in one end-point is not muted by another positive end-point component [82, 83].
Key considerations on the use of the 6MWT as a primary end-point, with areas for future research
Identification of suitable ILD patient populations, considering interstitial lung and pulmonary vascular components of the disease. Assessment of the degree of disease progression expected in this population during the study and how this might be affected by the study treatment.
Choice of appropriate end-point, including what component(s) of the 6MWT to use (e.g. 6MWD or oxygen desaturation) and what type of change to measure (e.g. categorical change or mean change per treatment group).
Should the 6MWT be part of composite end-point(s) that also include clinical worsening or clinical progression outcomes? Careful optimisation of a composite end-point is needed to ensure clinical relevance of the final outcome.
Should multiple components of the 6MWT (e.g. 6MWD, oxygen desaturation, heart rate recovery and/or dyspnoea) be combined to form a composite end-point (e.g. 10% decrease in 6MWD and 4% decrease in oxygen desaturation, or other possible combinations of 6MWT outcomes)?
How can the 6MWT be standardised (e.g. by specifying criteria for the management of supplementary oxygen use and stopping rules) to reduce variability of measurements and allow the comparison of findings from different trials?
Should a second 6MWT-related variable be used to adjudicate change in cases where change in the 6MWT-related primary end-point is marginal (as occurred in the trial of sildenafil added to pirfenidone in patients with advanced IPF and risk of PH [1])?
Conclusions
The 6MWT represents a “catch-all”, mirroring everyday exercise tolerance and status of individuals as a whole, which is not specific to ILD, pulmonary vasculature or extrapulmonary comorbidities. Since we believe that the 6MWT accurately measures global progression in advanced disease, we advocate for its use as a primary end-point selectively in advanced ILD and ILD-PH. Given the correlation between 6MWT-related outcomes and QoL, use of the 6MWT is of interest to patients, payers and healthcare providers, and is thus valuable and warranted to serve at minimum as a secondary end-point in ILD trials. We propose that a single 6MWT outcome is most suitable as a primary end-point in ILD trials if the treatment is expected to improve clinical performance or prevent rapid disease progression in the study population; in this situation, the 6MWT should be used as a continuous variable. Composite primary end-points, including 6MWT outcomes, could be combined to allow their use in any ILD population. Further research is needed to determine optimal use of the 6MWT as a primary end-point across ILD trials.
Acknowledgements
Medical writing support was provided by Rebekah Waters of CMC AFFINITY, McCann Health Medical Communications, UK, funded by F. Hoffmann-La Roche, Ltd.
Footnotes
Published in volume 31, issue 165 of the European Respiratory Review on 24 August 2022; republished 26 August 2022 with amendments to the authors' affiliation details.
Provenance: Submitted article, peer reviewed.
Data-sharing statement: Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://vivli.org). Further details on Roche's criteria for eligible studies are available here (https://vivli.org/members/ourmembers). For further details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, please see https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm
Author contributions: All authors contributed to the development of this review article from the outset and read and approved the final draft.
Conflict of interest: S. Harari has served as a consultant for, received speakers’ bureau fees from and received research funding from Actelion, Boehringer Ingelheim and F. Hoffmann-La Roche, Ltd, outside the submitted work.
Conflict of interest: A.U. Wells has received payments or honoraria and consulting fees to his institution from F. Hoffmann-La Roche, Ltd, and has received payments or honoraria and consulting fees both personally and to his institution from Boehringer Ingelheim, outside the submitted work.
Conflict of interest: W.A. Wuyts has received consulting and/or lecture fees from Boehringer Ingelheim and F. Hoffmann-La Roche, Ltd, outside the submitted work.
Conflict of interest: S.D. Nathan has served as a consultant for, received speakers’ bureau fees from and received research funding from Bellerophon, Boehringer Ingelheim, F. Hoffmann-La Roche, Ltd, Galapagos and United Therapeutics, outside the submitted work.
Conflict of interest: K-U. Kirchgaessler is an employee and shareholder of F. Hoffmann-La Roche, Ltd, disclosures made outside the submitted work.
Conflict of interest: M. Bengus is an employee and shareholder of F. Hoffmann-La Roche, Ltd, disclosures made outside the submitted work.
Conflict of interest: J. Behr reports sponsorship or research funds from BMBF, DFG and LMU-KUM, and has received payment or other financial remuneration from Actelion, AstraZeneca, Bayer, Biogen, BMS, Boehringer Ingelheim, F. Hoffmann-La Roche, Ltd, Galapagos, MSD, Novartis, Promedior and Sanofi/Genzyme, outside the submitted work.
Support statement: This review was funded by F. Hoffmann-La Roche, Ltd. Authors who are employees of F. Hoffmann-La Roche, Ltd were involved in the writing of the review manuscript in collaboration with the academic authors. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received May 6, 2022.
- Accepted June 14, 2022.
- Copyright ©The authors 2022
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References











Jump To
- Article
- Abstract
- Abstract
- Introduction
- Search strategy and selection criteria
- Conduct and use of the 6MWT in clinical practice
- Predictive factors for 6MWT outcomes and their prognostic value
- What 6MWT outcomes and DLCO tell us about ILD progression
- Use of the 6MWT as a primary end-point in clinical trials
- Proposed place of the 6MWT as a primary end-point in ILD trials
- Conclusions
- Acknowledgements
- Footnotes
- References
- Figures & Data
- Info & Metrics