





Abstract
Multidisciplinary team (MDT) meetings, involving the integrated collaboration of healthcare professionals, are increasingly used in clinical practice to inform the diagnosis and treatment of interstitial lung diseases (ILDs). Over time, the assessment of patients with ILD has transitioned from discussions among clinicians, radiologists and pathologists to the inclusion of a broader range of clinical data and specialist expertise. Studies have shown that a multidisciplinary approach can have many benefits for the clinical care of patients with ILD by improving the diagnostic confidence for different ILDs and guiding treatment decisions. The utility of MDT discussions for diagnosis, monitoring disease progression and management decisions, will need to be considered based on how it is best positioned in the diagnostic and therapeutic process, as well as the practicality and challenges of its use. There are also uncertainties and heterogeneity concerning the optimal practices of MDT meetings in ILD care. In this review, we describe recent developments refining the approach to MDTs in clinical practice, including who should be involved in the MDTs, when it is most needed, their use in patient management, challenges in their implementation, and ongoing controversies in the field that need further research.
Abstract
Due to the complexity of fibrotic ILDs, multidisciplinary teams are instrumental in optimising the diagnosis and management of patients. https://bit.ly/3R9pZ6R
Introduction
Interstitial lung diseases (ILDs) are heterogeneous disorders with different causes, clinical manifestations and treatment options. Idiopathic pulmonary fibrosis (IPF) is the most common fibrotic ILD and is characterised by a radiological and/or histological pattern of usual interstitial pneumonia (UIP) and progressive fibrosis [1]. Fibrotic ILDs are also associated with autoimmune and connective tissue diseases (CTDs), sarcoidosis and fibrotic hypersensitivity pneumonitis (HP). While fibrosis in these diseases may not always be progressive, a proportion of patients manifest progressive pulmonary fibrosis (PPF) [2], which shares a disease course similar to IPF and is characterised by high mortality and decline in lung function [3–5]. The similar clinical presentations of these varied conditions can result in difficulties obtaining a definitive diagnosis, which can impact clinical decision-making and the timely provision of appropriate treatments [1].
Early guidelines set by the American Thoracic Society (ATS)/European Respiratory Society (ERS) committee recommended the use of multidisciplinary teams (MDTs) as a gold standard for diagnosing patients with IPF [6, 7]. Subsequent expansion of these guidelines further strengthened the role of MDTs [8]. Several studies have shown that a comprehensive assessment of clinical, radiological and pathological data by ILD experts can improve diagnostic agreement [9–12]. MDTs also draw information from a range of specialities to interpret available evidence for reaching a consensus diagnosis [13]. While MDTs in ILD have mainly focussed on diagnosis, there is also evidence that MDT discussions may benefit management and follow-up of patients. Within other fields, such as oncology, MDTs are a key component in patient care and the treatment decision process. With the emergence of new therapeutic interventions being used to treat fibrotic ILDs, there is a need for more accurate diagnoses and better predictors of longitudinal disease behaviour in specific ILDs to further optimise and tailor individual patient treatment plans [14].
Multidisciplinary approaches may vary from one group to another, which may depend on the availability of local resources and differences in healthcare systems of different countries. In some settings, a multidisciplinary diagnosis of IPF is a condition for the reimbursement of antifibrotic treatments [15, 16]. Uncertainties regarding the optimal MDT structure remains a challenge [17]. Data comparing different approaches could help improve the framework around MDT practice and possibly lead to harmonisation of some of its aspects; however, there is a lack of formal criteria for the organisation and validation of MDTs [17].
MDTs are continually evolving in response to patient needs and therapeutic developments, and this review will outline current perspectives on their use in the clinical care of ILDs. A podcast summarising this review article is available at: www.globalmedcomms.com/respiratory/Cottin/MDTinongoingmanagementofILDs
History of MDTs in ILD
In 2002, a statement by the ATS and ERS advocated an integrated approach involving clinical, radiological and pathological data for the diagnosis of idiopathic interstitial pneumonia (IIP) [6]. A landmark study by Flaherty et al. [12] involving 58 patients with ILD showed that a multidisciplinary approach involving clinicians, radiologists and pathologists increased the confidence level for a given diagnosis. In this study, MDT participants were provided with information in a stepwise manner and asked to record their diagnostic impression and level of confidence when making a consensus diagnosis for patients with suspected IIP. This incremental process demonstrated that discussions and exchange of information between clinicians, radiologists and pathologists improved the overall interobserver agreement and confidence level for diagnosing ILD. In addition, histopathological findings led to improved agreement between clinicians and radiologists. This study underscored the importance of a multidisciplinary approach for improving the quality of how physicians diagnose ILD. Subsequently, MDTs have been increasingly recognised for diagnosing fibrotic ILDs in several consensus statements and guidelines that outline recommendations for their format and use (see table 1).
Guideline recommendations for the use of multidisciplinary teams (MDTs) in fibrotic interstitial lung diseases (ILDs)
In the past two decades, the longitudinal care needs of patients with ILD have resulted in specialist ILD centres to manage and support patients [18]. Results from a Delphi survey and patient focus group analysis focusing on the essential components of an ILD clinic have proposed multidisciplinary conferences as one of the main components [19]. In addition, the European Commission has established European reference networks to facilitate multidisciplinary discussions for rare lung diseases through networks involving healthcare providers across hospitals in Europe [20].
Disease characteristics and the roles within an MDT
Determining the aetiology of pulmonary fibrosis requires the comprehensive assessment of disease-specific factors, including a thorough history of extrapulmonary signs, exposures and medication use, serological testing, lung function testing, high-resolution computed tomography (HRCT) imaging and lung biopsy [1, 21]. Although genetic testing is not widely used for diagnosis, clinicians should routinely obtain family history of ILD and manifestations, as this may be suggestive of alterations in telomere-related genes [22]. Due to the complexity of disease presentation in cases of fibrotic ILDs, evaluating clinical evidence requires a multidisciplinary discussion involving expert specialists in the diagnostic pathway.
The ATS/ERS consensus statements and 2017 Fleischner Society white paper suggest that MDTs should, as a minimum, include a pulmonologist, a radiologist and a pathologist to integrate different clinical data before forming a final diagnosis [6, 7, 23]. The National Institute for Health and Care Excellence guidelines also stipulate a minimum composition of the MDT for diagnosing IPF, including a specialist radiologist, histopathologist, clinician and clinical nurse specialist [24]. A collective analysis of various studies by Furini et al. [13] found that the most frequently involved physicians in an MDT were thoracic pathologists (23/29 studies), thoracic radiologists (26/29 studies) and pulmonologists (29/29 studies). However, a range of other specialities was also reported in several studies, which include the integration of rheumatologists, clinical nurse specialists, occupational therapists, cardiothoracic surgeons and lung transplantation teams. The composition of members may depend on the purpose of an MDT (figure 1). It could be argued that the true purpose of an MDT in ILD is ultimately to make a diagnosis; as such, the core group of specialists would involve a pulmonologist and radiologist, with the inclusion of other specialities (pathologists and rheumatologists) considered on a case-by-case or centre-by-centre basis [25]. However, the scope of MDTs in ILD has expanded to encompass other specialists who may benefit the dynamic discussions around diagnosis, monitoring disease progression and management decisions. For example, rheumatologists and transplant surgeons are also key contributors in an MDT to provide management recommendations for CTD-ILDs and lung transplantation, respectively, although the availability of these speciality groups to participate in a meeting is a limitation [26]. Pulmonologists may be best placed to take part in monitoring and management discussions as they directly consult with the patient, along with integrating clinical data. Additional noncore members may also advise on the supportive care for patients (palliative care and transplant teams) but may not need to attend regular MDT meetings. Assessing patient comorbidities and preferences is especially important considering that treatment options are often associated with adverse events that may affect a patient's quality of life [26].
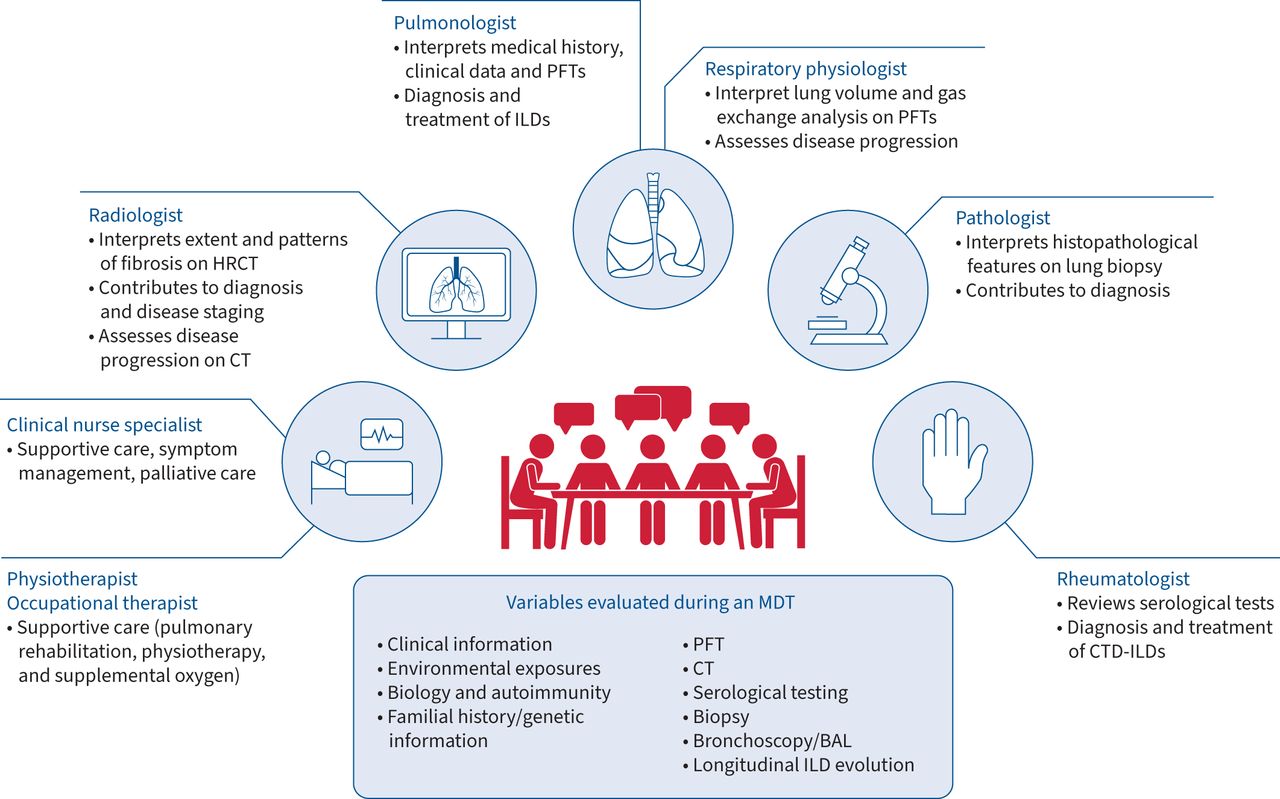
{kind=link}
Example of roles in an multidisciplinary team (MDT) for diagnosis and management. BAL: bronchoalveolar lavage; CT: computed tomography; CTD: connective tissue disease; HRCT: high-resolution computed tomography; ILD: interstitial lung disease; PFT: pulmonary function testing.
Regarding pathology, guidelines recommend that MDTs should first evaluate the available evidence collected based on typical clinical and HRCT features before evaluating whether a biopsy is necessary [7, 23]. If a biopsy is performed, a second MDT discussion may help to evaluate its results. It is important that the sample is adequately obtained to provide diagnostic information, and MDT discussions may be useful in determining the most appropriate area for tissue sampling and the type of biopsy used [16]. There is accumulating evidence that transbronchial lung cryobiopsy is a safe method for histopathological diagnosis. The decision to use it over surgical lung biopsy is evolving, taking into account the institutional expertise of the operator and the pathologist, with recent ATS guidelines recommending its use for evaluating fibrotic HP and IPF [2, 27–29]. In this regard, the multidisciplinary discussion integrates the available information, including local experience with different techniques and their local availability, to propose the best approach to diagnosis.
The involvement of a rheumatologist is crucial in the assessment of CTD-ILDs [15] but may not be mandatory in every ILD MDT discussion. In the care pathway of a rare CTD, if a patient develops a manifestation of ILD during an established CTD, rheumatologists are commonly involved. On the other hand, if the pulmonary involvement is the first manifestation, with CTD symptoms appearing later, the patient will logically be discussed in an ILD MDT, where the pulmonologist will convene with a rheumatologist. Current guidelines suggest that rheumatologists could be involved on a case-by-case basis, and patients should be referred to a rheumatologist when showing positivity from serological tests (e.g. autoantibody tests) or presenting clinical manifestations suggesting an underlying rheumatological disease [8]. In some cases, it may be difficult to distinguish patients with IIP from those with CTD-ILD, as with interstitial pneumonia with autoimmune features, which does not fit classical CTD criteria [13]. Rheumatology input may be particularly valuable during the examination of the patient and history taking, where subtle signs of a CTD may be elicited that might otherwise be missed [30, 31]. Additionally, the rheumatologist may aid in management decisions for rare CTDs with a heterogeneous course of associated ILD [32, 33].
What cases are likely to benefit from MDT characterisation?
It should be noted that not all cases of suspected ILD require MDT characterisation. Since the initial clinical assessment can determine a diagnosis of IPF based on radiological evidence of UIP and clinical history, or confirm CTD-ILD through computed tomography and serological testing, an MDT may not always be required. Given the risks of additional invasive procedures, patients for whom this approach is being considered may benefit from MDT discussion before and after a biopsy. Therefore, an MDT would be needed in only a proportion of cases with difficult diagnostic and therapeutic dilemmas.
A proportion of ILDs remains unclassifiable after initial assessment [34]. Methods to classify different types of fibrotic ILD include terminology based on diagnostic confidence (confident diagnosis, provisional diagnosis and unclassifiable), as suggested by a working group led by Ryerson et al. [35]. One of the roles of an MDT is to assess the level of confidence in the diagnosis, to propose a first-choice diagnosis and to identify alternative diagnoses that can be considered. When there is no clear diagnosis, patients could be managed based on a “working diagnosis” – a non-definite diagnosis based on sufficient confidence and clinical reasoning. This accounts for disease behaviour over time, potentially requiring future MDTs at regular intervals to evaluate clinical information and HRCT evolution [30, 36, 37]. Recently, a Bayesian diagnostic approach has been proposed to integrate the clinician's clinical judgement into an IPF diagnosis, where the clinical features and the disease behaviour (e.g. observed outcome of disease with or without therapy) are integrated together with radiological and pathological features when available [37]. This concept especially helps to differentiate IPF from other fibrotic ILDs and may obviate the need for histopathology in a number of cases.
The presence of a UIP pattern on HRCT is a key component in the diagnostic process of IPF. In patients with a clear UIP pattern and no signs of antigen, environmental exposure or a systemic disorder, a diagnosis of IPF may not require an MDT [7]. Guidelines suggest that an MDT can be especially relevant in cases where the radiological pattern is not UIP, and when it is discordant from the pathological pattern (e.g. HRCT is inconsistent with UIP, but the histopathology result indicates UIP) [38].
Although UIP is commonly associated with IPF, it is also observed in other ILDs, such as fibrotic HP and rheumatoid arthritis-associated ILD [39]. Patients with fibrotic HP can have similar histopathological and radiological manifestations to those with IPF, including a UIP-like pattern of fibrosis on lung biopsy, requiring the assessment of exposure-related symptoms and HRCT features to distinguish fibrotic HP from IPF [29, 40]. A potential exogenous origin may still be causative in the case of definite UIP, and a careful assessment for exposure (history, standardised questionnaire, serology) and bronchoalveolar lavage may be instructive [27, 29, 41]. The risks and invasiveness of performing a biopsy can deter patients from its use when it is needed for the confirmation of a UIP pattern. In the absence of biopsy, key multidisciplinary information that can impact the “classic UIP” found on HRCT includes age, gender, decline in lung function and smoking history, which can determine a working diagnosis of IPF or provide an alternative diagnosis of a different ILD [42, 43]. In some cases, patients initially considered not to have IPF who fail to respond to immunosuppressive treatment experience an irreversible, fibrosing phenotype of disease progression, leading clinicians to revisit the diagnosis and reconsider the diagnosis of IPF in an MDT discussion based on disease behaviour.
Multiple groups have highlighted the effect of MDT discussion for confirming or changing the preliminary diagnosis [42, 44]. De Sadeleer et al. [45] demonstrated that an MDT changed the diagnosis in 41.9% of patients, as well as suggesting first-choice diagnosis in 80.5% of cases previously considered unclassifiable. In another study, ILD diagnoses were changed in 53% of patients following MDT discussion, resulting in an increase in the proportion of patients diagnosed with CTD-ILD (from 10% to 21%) and hypersensitivity pneumonitis (from 3% to 16%) [46]. Overall, this highlights that as well as their role in diagnosing IPF, MDTs play a role in the broader discussion around non-IPF ILD and can help expand the expertise and confidence for making an alternative diagnosis.
Impact of MDTs on the management of fibrotic ILDs
Accuracy of diagnosis on management
Several studies have investigated the potential benefits of MDT diagnosis on improving inter-observer agreement or leading to a change in the final diagnosis or management recommendation using a “pre-MDT” and “post-MDT” study design (table 2). The accuracy of MDT diagnosis of ILD is an important step that leads to the development of management strategies for specific ILDs, especially where the underlying cause of the disease is rare and difficult to assess [17]. For example, identifying inciting antigens can improve outcomes for patients diagnosed with fibrotic HP [47]. A global study evaluating ILD diagnostic practices across different countries found that patient management was discussed in 94.9% of MDT diagnostic meetings in centres [48].
Summary of the impact of multidisciplinary team (MDT) discussion in interstitial lung disease (ILD) diagnosis and treatment
The distinction between IPF and non-IPF ILDs is particularly important given the worse prognosis in IPF compared with other fibrotic ILDs. Being able to distinguish between IPF and non-IPF ILDs is critical for clinical practice, where immunosuppression may benefit non-IPF ILDs but is associated with increased risk of death and hospitalisation for patients with IPF [49, 50]. Studies have shown that management recommendations following MDT discussion can lead to changes in the use of corticosteroids and immunosuppressive therapies, nonpharmacological therapies (e.g. supplemental oxygen), pulmonary vasodilators and antifibrotics [15, 46], highlighting the impact that MDTs can have on the treatment of patients with suspected ILD.
Monitoring and management of progressive disease
Different subtypes within the spectrum of fibrotic ILDs can develop PPF despite immunosuppressive therapy and the avoidance of disease-triggering stimuli. Patients with different ILDs have variable trajectories for progression, with some remaining stable and others developing inexorable progression [1]. As studies have shown that IPF and PPF may share similar pathophysiological mechanisms, it has been proposed that patients should be grouped for treatment based on shared disease behaviour, regardless of the underlying diagnosis [51]. This concept has been supported in the 2013 ATS/ERS classification, highlighting that management should be based on the most probable diagnosis after MDT discussion and consideration of expected disease behaviour [7]. Data from the INBUILD trial showed that patients with PPF other than IPF showed similar disease progression in terms of decline in forced vital capacity and mortality compared with patients with IPF in the INPULSIS trials [52]. Therefore, the correct diagnosis and follow-up of patients are needed to better define progression in fibrotic ILDs, which can have implications for therapeutic strategy [53]. Unfortunately, there has been no standardised definition for progression, and it may be difficult to predict when fibrotic ILDs will become progressive. The recent 2022 international guideline has included a definition of PPF for use in clinical practice, based on deteriorating lung function, HRCT findings and patient symptoms [2]. As disease progression can be monitored using a variety of methods, MDTs may be useful in the follow-up of patients with ILD [54]. The input of pulmonologists and serial pulmonary function tests (PFTs) is one of the main components for monitoring disease progression, and it has previously been shown that a decline in forced vital capacity of ≥10% is associated with greater mortality [55, 56]. In many university hospitals, running the PFT lab has become a subspeciality of pulmonology/physiology, with respiratory physiologists playing an increasing role in patient management through their contribution in interpreting pulmonary physiology variables and assessing disease progression. Other than lung function, the multifaceted approach for monitoring may include a combination of HRCT imaging, exercise testing and symptom evaluation [1, 57]. Serial HRCT monitoring has been evaluated in a number of studies on its ability to provide information on the evolution of ILD in patients with worsening symptoms [58]. Wider use of quantitative imaging techniques, to more accurately characterise the extent of lung fibrosis to supplement the visual assessment by radiologists, could lead to earlier identification of disease progression during routine HRCT follow-up [58, 59].
As the disease course is continually monitored, the role of MDT discussion is no longer simply a diagnostic exercise, but a discussion of the ongoing management options and evidence of ILD progression (see table 3). Based on the results of clinical trials, both nintedanib and pirfenidone were able to slow the decline in lung function compared with placebo in patients with non-IPF fibrotic ILDs [60, 61], as well as unclassifiable ILD [62]. The sequence of medications is important, as the use of immunosuppressive treatment is usually considered as first-line therapy, whereas antifibrotic treatment is initiated to manage progressive disease. In rheumatoid arthritis-associated ILD, given the need to treat both the ILD and articular disease, it has been suggested that multidisciplinary decision-making involving both pulmonologists and rheumatologists is the optimal model for making treatment decisions [63]. This can help to determine whether treatment goals should be driven by systemic disease, pulmonary disease, or both, particularly in the case of cardiopulmonary disorder co-existing with systemic disease [64]. The optimal therapeutic strategy for systemic sclerosis (SSc)-associated ILD involves the consideration of antifibrotic treatment alone or in the background of immunosuppressive therapy (primarily mycophenolate) or no therapy, based on evidence of it preserving lung function in the SENSCIS (Safety and Efficacy of Nintedanib in Systemic Sclerosis) trial in SSc-ILD [65, 66]. More recently, the anti-interleukin-6 receptor antibody tocilizumab has been approved for the treatment of SSc-ILD [67]. Given that, in some cases, patients that present with CTD cases may be assessed by a rare CTD MDT (rheumatologist, pulmonologist, cardiologist, radiologist), it is important to refer to the multidisciplinary specialised ILD clinic when appropriate.
Use of multidisciplinary team (MDTs) in diagnosis, monitoring and management
Supportive care and lung transplantation
At a multidisciplinary specialised ILD clinic, patients are supported by a network of professionals, including specialist nurses, physiotherapists and expert physicians, as well as given access to restricted medication and enrolment into clinical trials [18]. A study evaluating factors that patients considered important in attending an ILD clinic showed that 86% valued a multidisciplinary approach in their care [68].
Beyond the initial diagnosis, MDTs can be important in supportive care by helping patients further understand their disease and allowing patients and families to make decisions in end-of-life planning. In one outpatient study, a team of respiratory care experts, nurses, respiratory therapists, physiotherapists and a dietitian helped to reduce hospitalisations by adopting an early integrated palliative approach, with a focus on early symptom management and advance care planning [69]. Previous studies investigating the unmet needs and preferences of patients and healthcare professionals have highlighted the importance of shared care and multidisciplinary collaboration in management [70–72]. A multidisciplinary approach is also considered key for lung transplantation, as a discussion between clinicians and the transplant team can help to improve outcomes before the surgery takes place [73].
Important considerations for implementing MDTs in clinical practice
There is a need to understand best practices of MDTs to implement them in the clinical setting. Previous diagnostic guidelines and surveys have shown that there is considerable heterogeneity in MDT meeting practices [25], and there are still unresolved controversies concerning MDT practices that will need to be addressed in future studies (table 4). A recent Delphi study was conducted among ILD experts to gain consensus on the essential and desirable features of an ILD MDT meeting [74]. Obtaining high-quality HRCT scans and the presence of a radiologist were highlighted as essential, whereas a pathologist was highly desirable. Although the role of MDTs in management recommendations was considered highly desirable, this was controversial since treatment depends on individual patient factors such as frailty and personal wishes, which cannot be understood through a discussion among ILD specialists [74]. As a minimum requirement, we recommend that cases are discussed in an MDT involving at least a pulmonologist and radiologist. MDTs should discuss difficult diagnostic cases and consider the need for a biopsy, as well as the type of biopsy (cryobiopsy/surgical lung biopsy). In this setting, the participation of a pathologist or the individual to perform invasive sampling may provide key input in identifying the site of sampling. Post-biopsy results will also need to be considered in a further MDT discussion to determine the diagnosis, which can provide the basis for therapeutic decisions.
Unresolved controversies regarding multidisciplinary team (MDT) practice
Consistent levels of agreement are not always possible through an MDT discussion. Poor inter-MDT agreement was found between different centres in diagnosing HP and nonspecific interstitial pneumonia, which may be related to the lack of diagnostic criteria for these ILD subtypes [10]. Further research is needed on shared practices among ILD MDTs, including the format of meetings, documentation of cases and meeting coordination, to reduce heterogeneity and allow for inter-MDT benchmarking [26, 74]. A standardised template of collated patient data and regular multidisciplinary conferences (ideally once every 2 weeks) can increase harmonisation and maintain expertise [19, 26, 74]. Through a multidisciplinary discussion between team members, a collaborative environment is established within which each member contributes their expertise, which can help to improve the skillset and experience of the involved clinicians. Regular attendance at MDT meetings was shown to improve the prognostic accuracy of nonacademic clinicians up to the level of experienced ILD experts [75]. In other specialities, research into the quality of group decision-making has highlighted key factors for effective teams, such as meeting management, training, leadership, teamwork and culture, and incorporating patient views [76, 77]. Understanding how the dynamics of participants within an MDT may influence decisions and the impact of other factors will help to develop strategies for effective MDTs in the care pathway for fibrotic ILDs.
There is a lack of methods to evaluate the performance of an MDT, and difficulties in designing studies to assess its characteristics. There is currently no independent reference standard to validate an MDT diagnosis against since all the diagnostic information is considered in an MDT. It has been proposed that multidisciplinary diagnosis of IPF can be validated against the course of the disease by comparing outcomes between IPF and non-IPF diagnoses [10, 75, 78]. In one study by Walsh et al. [10], by using outcome distinctions between IPF and other ILDS as a surrogate for diagnostic accuracy, MDT diagnosis of IPF was more prognostically accurate than IPF diagnoses made by the individual specialists of the MDT. Assessing whether management recommendations and treatment plans are being followed by patients is another potential indicator for the effectiveness of MDTs [77].
Barriers to implementation
Not all institutions are able to have an MDT in place, and expert centres are currently the primary location for multidisciplinary assessment [18]. While MDTs facilitate a common language of communication between the specialists, the quality of an MDT may depend on the experience and expertise of the individual physicians that form it and the accuracy of the clinical information gathered [79]. The complexity of the process in diagnosing ILD, such as detecting signs of CTD-ILD and identifying environmental exposures in fibrotic HP, suggests a need for members experienced in ILD to maximise the quality of interpreting clinical data in individual patients. Other hurdles in implementing an MDT include the potential that interactions within an MDT at a personal level may create unbalanced discussions, especially when the skills and expertise of the specialists involved differ significantly. A lack of common goals, uneven levels of expertise and inexperience with technology are some of the challenges that limit integrated collaboration of MDT members [17, 80].
A review of multidisciplinary meeting practices in expert ILD centres by Jo et al. [46] demonstrated significant heterogeneity in terms of meeting format and organisation, suggesting that physicians should adapt to local organisations and constraints to facilitate a good interaction between speciality physicians. This article reported the results of a survey from 10 expert centres and showed that the attendant clinician was responsible for leading the meeting in 90% of the centres, documenting the outcome of the meeting 70% of the time, and had the greatest input into formulating diagnosis 60% of the time [81]. In an international multicentre study of ILD MDTs, pulmonology was almost always represented (99.7% of centres), with radiology present at most centres (91.4%); however, histopathology specialist attendance was less frequent, with approximately a third of centres (including 26% of academic ILD centres) having no regular pathologist participant. This suggests that a low proportion of cases used biopsies to establish a diagnosis [48]. The reason for the lack of pathologist participation is not clear: this may have been due to the unavailability of biopsies and/or the few cases in which they would be required to contribute. In the same study, it was found that formal MDT meetings were more likely to be held at ILD academic centres than non-ILD academic centres or nonacademic centres, and were more common in centres in countries with higher income [48].
The process of running an MDT in busy academic practices can be burdensome and time-consuming for its participants. Where this is not possible, the treating physician should assimilate the multidisciplinary factors and evidence in each case before making diagnostic and therapeutic decisions [1]. In addition, a “nonmultidisciplinary” discussion between pulmonologists, which may take place in or outside the MDT, may be important in determining management decisions and treatment options, especially for clinicians less experienced in ILD. Smaller, community-based practices may benefit most from MDT characterisation for their cases but may lack ILD experience in their group participants. Telemedicine may be an option to allow greater access to MDTs in clinical practice and could create opportunities for improved communication when diagnosing and managing patients [82]. Virtual meetings between experts and community groups can help address gaps in expertise. The use of telemedicine has been accelerated during the coronavirus disease 2019 (COVID-19) pandemic, as it provides a platform for discussion and communication despite the geographic separation of ILD experts and limited healthcare resources. However, it should be noted that telemedicine has been applied in MDT meetings before the COVID-19 pandemic, as services across Australia have utilised a hybrid virtual/in-person MDT approach in their clinical practice [83]. Telemedicine also allows the use of enhanced and secure data-sharing platforms, which can increase collaboration among specialist and general practitioners. In one study, a web-based MDT with specialists in pulmonology, radiology and pathology was conducted for 465 cases of biopsy-proven IIP, using a nationwide cloud-based integrated database and video conferencing [82]. Although the MDT format was feasible and led to better prognostic discrimination among IIPs, it is important to note that the quality of the history and physical examination available in the database may depend on the experience of each patient's attending physician in managing ILD. Patient confidentiality should also be preserved during virtual case presentation [83]. Grewal et al. [84] reported findings on the feasibility of MDTs for evaluating the diagnosis and management of 91 external patients remotely. The results indicated that remote MDTs could be effective in changing the diagnosis of external patients and, in a survey, the use of external review was considered satisfactory by 93% of referring pulmonologists. However, the efficacy of remote MDTs will need to be balanced with the benefits derived from traditional in-person assessments within ILD clinics, which include access to additional support from specialist nurses, support groups, patient educators and counselling [84].
Conclusion
MDTs can play an important role in improving the quality of how physicians make diagnostic and management decisions. The practicality of running an MDT, determining when it is most needed, and its standardisation are still matters of debate. MDTs can provide significant cooperation and alignment on diagnostic evidence to support the final diagnosis of a complex case, particularly where evidence and standardised diagnostic guidelines may be lacking. Previous studies have highlighted their ability to increase interobserver agreement and establish an accurate diagnosis, which can benefit the prognostication of ILD disease course.
Although the MDT was originally developed to improve diagnostic evaluation of ILD, discussions have also turned to strategies around management to optimise patient outcomes. Considering the need to re-review patients who progress despite therapy, and the impact of an alternative diagnosis, an MDT approach is here and available to help the clinician assess whether disease progression has been evidenced and warrants management decisions, or to help discuss challenging treatment decisions.
Further studies of MDT approaches will be needed to address how they can be successfully utilised in clinical practice.
Footnotes
Provenance: Submitted article, peer reviewed.
Conflicts of interest: V. Cottin reports grants from Boehringer Ingelheim; consulting fees from Boehringer Ingelheim, Roche, Galapagos, Galecto, Shionogi, Fibrogen, RedX and PureTech; member of trial event adjudication committee for Fibrogen; payment for lectures/presentations/speakers bureaus/manuscript writing/educational events from Boehringer Ingelheim and Roche; travel support from Boehringer Ingelheim and Roche; and participation in data safety monitoring boards from Roche/Promedior, Celgene/BMS and Galapagos. F.J. Martinez has served on a steering committee, advisory board, data safety monitoring board, or adjudication committee for Afferent/Merck, Bayer, Biogen, Boehringer Ingelheim, Nitto, Respivant, Roche and Veracyte; has received consulting fees or payment for presentations from AbbVie, Boehringer Ingelheim, Bristol-Myers Squibb, Bridge Biotherapeutics, CSL Behring, DevPro, IQVIA, Roche/Genentech, Sanofi, Shionogi, twoXAR, United Therapeutics and Vercyte. V. Smith reports grants from Boehringer Ingelheim, Janssen-Cilag, Research Foundation Flanders and Belgian Fund for Scientific Research in Rheumatic Diseases; has served as a consultant/speaker for Boehringer Ingelheim, Actelion, UCB, Galapagos, Accord Healthcare and Janssen-Cilag; reports support for attending meetings and/or travel from Celgene and Boehringer Ingelheim; and reports leadership or fiduciary roles for EULAR Study group on Microcirculation in Rheumatic Diseases (Chair, unpaid), ACR Study Group on Microcirculation (Co-chair, unpaid), SCTC working group on capillaroscopie (Co-chair, unpaid) and ERN-ReCONNET (Steering committee member, unpaid). S.L.F. Walsh discloses relationships with Boehringer Ingelheim, Bracco, FLUIDDA, Galapagos, OncoArendi Therapeutics, The Open Source Imaging Consortium, Roche and Sanofi-Genzyme.
Support statement: The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). The authors did not receive payment related to the development of the manuscript. Darren Chow, MSc, of Meditech Media, UK, provided writing, editorial support, and formatting assistance, which was contracted and funded by Boehringer Ingelheim International GmbH (BI). BI was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received January 6, 2022.
- Accepted July 4, 2022.
- Copyright ©The authors 2022
This version is distributed under the terms of the Creative Commons Attribution Licence 4.0.
References











Jump To
- Article
- Abstract
- Abstract
- Introduction
- History of MDTs in ILD
- Disease characteristics and the roles within an MDT
- What cases are likely to benefit from MDT characterisation?
- Impact of MDTs on the management of fibrotic ILDs
- Important considerations for implementing MDTs in clinical practice
- Conclusion
- Footnotes
- References
- Figures & Data
- Info & Metrics