





Abstract
Pulmonary hypertension (PH) is known to complicate various forms of interstitial lung disease (ILD), including idiopathic pulmonary fibrosis, the interstitial pneumonias and chronic hypersensitivity pneumonitis. Pathogenesis of PH-ILD remains incompletely understood, and probably has overlap with other forms of pre-capillary pulmonary hypertension. PH-ILD carries a poor prognosis, and is associated with increased oxygen requirements, and a decline in functional capacity and exercise tolerance. Despite most patients having mild–moderate pulmonary hypertension, more severe pulmonary hypertension and signs of right heart failure are observed in a subset of cases. Clinical suspicion and findings on pulmonary function, computed tomography and echocardiography are often the initial steps towards diagnosis. Definitive diagnosis is obtained by right heart catheterisation demonstrating pre-capillary pulmonary hypertension. Drugs approved for pulmonary arterial hypertension have been investigated in several randomised controlled trials in PH-ILD patients, leading to discouraging results until the recent INCREASE study. This review provides an overview of the current understanding, approach to diagnosis and recent advances in treatment.
Abstract
Pulmonary hypertension complicates various forms of ILD and carries a poor prognosis. Diagnosis requires clinical suspicion and right heart catheterisation. Treatment remains limited, although results of the INCREASE study show promise. https://bit.ly/3mBokJv
Introduction
Interstitial lung diseases (ILDs) including idiopathic pulmonary fibrosis (IPF) and ILD associated with connective tissue disease may be complicated by pulmonary hypertension (PH-ILD). According to the World Symposium on Pulmonary Hypertension (WSPH), pulmonary hypertension group-3 clinical classification includes pulmonary hypertension due to lung diseases and/or hypoxia (figure 1). Pulmonary hypertension has been described in up to 86% of patients affected by ILD and is generally mild to moderate [1–3]. In this review we discuss the approach to diagnosis and the importance of identifying patients with PH-ILD early. We review the recent clinical trials and the evidence available to guide the treatment and management of PH-ILD.
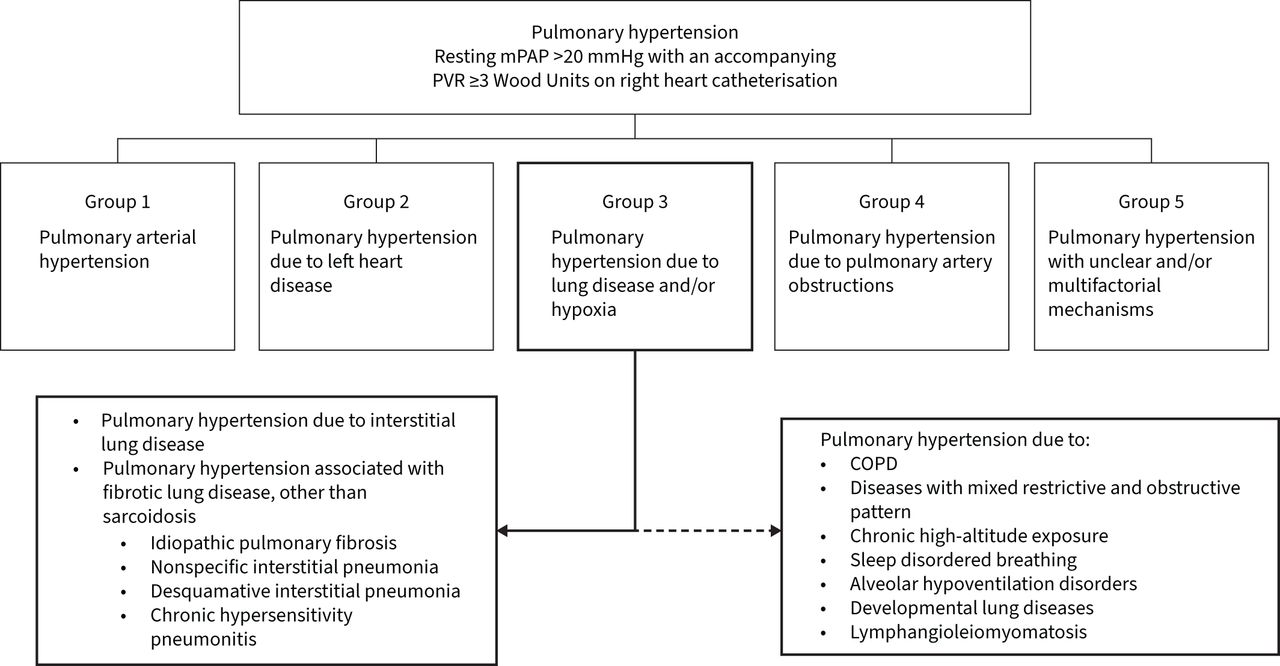
World Symposium on Pulmonary Hypertension classification of pulmonary hypertension. mPAP: mean pulmonary artery pressure; PVR: pulmonary vascular resistance.
Aetiology and classification
The diagnosis of ILD is often delayed and all too frequently recognised only at an advanced stage of the disease [4–7]. Aetiologies of the various ILDs are broad and include, although are not limited to, idiopathic, inhalational and immunological causes [8]. Idiopathic interstitial pneumonias (IIPs) are the most common, of which IPF is the most frequent subtype. This group includes nonspecific interstitial pneumonitis (NSIP), desquamative interstitial pneumonitis and usual interstitial pneumonitis. IIPs are distinguished by distinctive radiographic and pathologic features. Other ILDs include environmental and occupational diseases including chronic hypersensitivity pneumonitis; multisystem diseases including connective tissue disease and sarcoidosis; and combined pulmonary fibrosis and emphysema. Unusual ILDs include pulmonary Langerhans cell histiocytosis (PLCH) and lymphangioleiomyomatosis (LAM).
While various tools have been used to assess pulmonary hypertension, right heart catheterisation (RHC) still represents the gold standard for diagnosis [3, 9, 10]. Unfortunately, this evaluation is generally performed in patients after referral for lung transplantation, so data from several studies have been collected in this setting, which is generally represented by younger subjects with severe disease, but without significant medical comorbidities [11].
In a significant percentage of patients, pulmonary artery pressures and resistance values are disproportionately increased, suggesting vascular remodelling that is not simply a result of hypoxia and parenchymal injury. The pathogenesis of this phenomenon remains unclear [9, 11–13]. The pathogenesis of PH-ILD has features that overlap with what is known in other forms of pulmonary vascular remodelling, including WSPH group-1 pulmonary arterial hypertension (PAH). With diffuse parenchymal lung disease there is a role for local compression of pulmonary arteries. Hypoxic pulmonary vasoconstriction (HVC) is a teleological protective mechanism that diverts blood flow from diseased regions of lung and occurs in response to alveolar hypoxaemia. Repeated or sustained periods of HVC can lead to pulmonary vascular remodelling and pulmonary hypertension [14]. Pathologically, pulmonary arterial vasculopathic changes like those seen in group-1 PAH, including adventitial changes, smooth muscle cell hypertrophy and intimal remodelling are seen within the parenchymal changes that differentiate the various diseases [15]. Potential causative factors include in situ thrombosis, a disrupted inflammatory background that results in release of various inflammatory and pro-proliferative cytokines, and vascular bed obliteration with vessel distortion caused by parenchymal destruction by ongoing interstitial inflammation and fibrosis [16, 17]. Pulmonary endothelial dysfunction is probably driven by mediators that are common to group-1 disease including platelet-derived growth factor, transforming growth factor-β, endothelin-1 and tumour necrosis factor-α [18–20].
In patients affected by combined pulmonary fibrosis and emphysema (CPFE), severe pulmonary hypertension has frequently been observed in 47–90% of patients [21, 22]. Although the pathogenic mechanisms underlying this phenomenon are still not well elucidated, a reduction of the capillary bed secondary to fibrosis and associated hypoxic pulmonary vasoconstriction have been suggested.
Pulmonary hypertension may be a clinical manifestation associated with other lung diseases, such as PLCH, LAM and sarcoidosis. The development of pulmonary hypertension in this group of diseases is associated with a worse prognosis and an accelerated decline in exercise performance. Often, supplemental oxygen is required [21–23].
Previously, pulmonary hypertension associated with these nonfibrotic ILDs was included in group 5 of the WSPH pulmonary hypertension classification, as a multifactorial pathogenesis was hypothesised. But in the most recent WSPH classification, LAM is considered part of group 3.
The presence of pulmonary hypertension was described for the first time in a series of patients affected by PLCH and confirmed in several reports [22, 24–27]. Several pathogenetic mechanisms have been hypothesised, such as pulmonary vasculitis involving arterioles and venules and vascular remodelling observed in patients with idiopathic PAH and inflammation, which are also commonly observed in patients with various other types of pulmonary hypertension [21]. Pulmonary hypertension is rarer in LAM patients and generally less severe compared to that observed with PLCH, although both are cystic lung diseases [28]. Regarding sarcoidosis, the severity of pulmonary function is not related to the development of pulmonary hypertension, and different mechanisms, such as sarcoid granulomatous vasculitis, mechanical compression of the large pulmonary arteries by adenopathy and distortion of the vascular bed, and fibrous mediastinitis or portal hypertension are supposed to play a role in the pathogenesis of pulmonary hypertension in this group of patients [29].
Haemodynamic classification
In 2018, during the 6th WSPH, the threshold of mean pulmonary artery pressure (mPAP) for the definition of pulmonary hypertension was reduced to 20 mmHg from 25 mmHg, representing an important change [30]. Pulmonary hypertension due to chronic respiratory diseases is now haemodynamically defined by mPAP >20 mmHg, a pulmonary artery wedge pressure ≤15 mmHg and an elevated pulmonary vascular resistance (PVR) ≥3 Wood Units [30]. The outcome of PH-ILD patients with mPAP values from 20 to 25 mmHg has still not been well evaluated; however, it is expected that this change could have an impact in terms of increased diagnoses [10, 31, 32]. Even the haemodynamic classification of pulmonary hypertension in the context of chronic lung disease has been modified. In fact, pulmonary hypertension with values of mPAP 21–24 mmHg and PVR ≥3 Wood Units or mPAP 25–34 mmHg was considered as mild–moderate, while severe pulmonary hypertension was defined in presence of mPAP ≥35 mmHg or mPAP ≥25 mmHg associated with a cardiac index <2 L·min−1·m−2 [33].
In some cases, the development of pulmonary hypertension may be unrelated to the underlying pulmonary disease and a concomitant presence of PAH and ILD could be hypothesised. This sort of “disproportionate” pulmonary hypertension in ILD should be suspected if the presence of severe pulmonary hypertension is detected in patients with a normal or mild respiratory function impairment (forced vital capacity (FVC) >70%) and no gross parenchymal involvement on computed tomographic imaging. This group of patients should be referred to an expert centre and evaluated using RHC. Treatment with drugs approved for group 1 pulmonary hypertension could be effective in subjects with a predominant pulmonary vascular involvement, rather than simple vascular rarefaction.
Screening and diagnostic evaluation
Although PH-ILD is more frequent in patients with severe underlying pulmonary parenchymal fibrosis, it may occur in milder disease. Because pulmonary hypertension negatively affects prognosis and quality of life in PH-ILD, screening for pulmonary hypertension in ILD, and in particular in IPF, is essential for a prompt diagnosis.
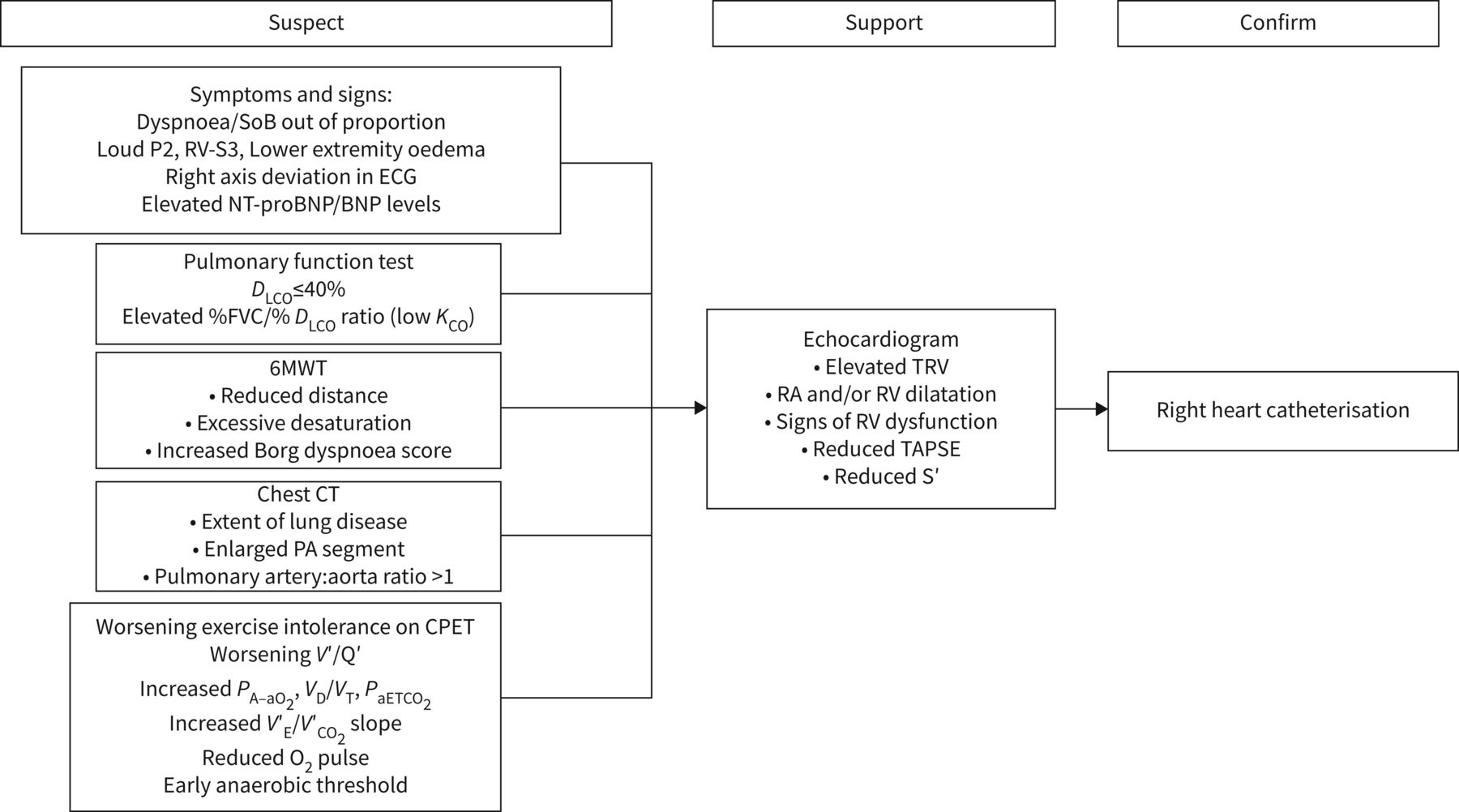
The presence of pulmonary hypertension should be suspected when patients complain of symptoms of worsening dyspnoea and fatigue and have a drop in diffusing capacity that is out of proportion to their imaging or pulmonary function findings. It should also be suspected in cases with signs of right heart failure (figure 2). As with any patient with ILD and worsening symptoms, it is necessary to be vigilant and assess for more common causes including venous thromboembolism and chronic thromboembolic disease, exacerbation of underlying lung disease, left heart disease including progressive valvular disease, and infection.

{kind=link}
{kind=link}
Algorithm for diagnosis of pulmonary hypertension. SoB: shortness of breath; ECG: electrocardiogram; NT-proBNP: N-terminal pro-brain natriuretic peptide; DLCO: diffusing capacity of the lung for carbon monoxide; FVC: forced vital capacity; KCO: transfer coefficient of the lung for carbon monoxide; 6MWT: 6-min walk test; CT: computed tomography; PA: pulmonary artery; CPET: cardiopulmonary exercise testing; V′/Q′: ventilation/perfusion ratio; PA–aO2: alveolar–arterial oxygen tension difference; VD: dead space volume; VT: tidal volume; PaETCO2: end-tidal arterial carbon dioxide tension; V′E: minute ventilation; V′CO2: carbon dioxide production; O2: oxygen; TRV: tricuspid regurgitant velocity; RA: right atrium; RV: right ventricle; TAPSE: tricuspid annular plane systolic excursion; S′: systolic velocity.
The role of pulmonary function testing
In the management of ILD patients, serial pulmonary function tests (PFTs) are useful for assessing the severity and progression of disease. Finding appropriate PFT values to suspect pulmonary hypertension in this group of patients could be valuable. However, this can be difficult because FVC, generally used to assess ILD severity, is less useful in pulmonary vascular diseases, and both vascular and fibrotic diseases lead to a reduction in the diffusing capacity for carbon monoxide (DLCO). In patients affected by PH-IPF who underwent RHC during a pre-lung transplantation evaluation, DLCO was lower compared to IPF subjects without pulmonary hypertension [9]. Interestingly, a combination of DLCO <40% and the necessity for oxygen supplementation were specific, but not sensitive, for identifying pulmonary hypertension in this population. Patients meeting these criteria had a positive predictive value of 86.7% for being diagnosed with pulmonary hypertension. Conversely, patients with none of these criteria still had a ∼20% chance [9].
According to the model of Roughton and Forster [34], two conductances arranged in series, the membrane conductance for carbon monoxide (DmCO) and the carbon monoxide loading on haemoglobin, which is the product of carboxyhaemoglobin and the mass of haemoglobin in alveolar capillary blood volume (Vcap), comprise the DLCO. DmCO reflects the characteristics of the alveolar capillary membrane, while Vcap is associated with the amount of blood volume in the ventilated alveoli. The measurement of diffusing capacity of the lung using nitric oxide (DLNO) along with DLCO allows the values of DmCO and Vcap to be obtained in a single-breath test. The clinical relevance of combined DLCO/DLNO in patients affected by ILD to predict the development of pulmonary hypertension has not yet been well elucidated. In a group of ILD patients (12 IPF and 10 NSIP), DmCO and Vcap were severely and homogeneously reduced, even with disease progression, regardless of whether pulmonary hypertension was present or not. Furthermore, in the same study, systolic pulmonary arterial pressure (sPAP) was negatively related to DmCO and Vcap [35]. These findings suggest that in ILD the decrease in DLCO is reduced by an increased thickening of the membrane as well as by a reduction in, or compression of, capillaries, as observed in the lungs of patients who died during an acute exacerbation [36]. Conversely, in a study involving patients affected by different ILDs, Vcap was often high or normal in the subgroup with associated pulmonary hypertension [33]. Sivova et al. [37] observed in patients affected by systemic sclerosis that Vcap and DmCO were reduced in ILD-PH group, but Vcap had the highest sensitivity and specificity in detecting PAH in patients with systemic sclerosis (SSc) with associated ILD. Additionally, in SSc patients, it has been suggested that FVC/DLCO (% pred) ratio >1.6 might be useful in predicting PAH, regardless of the extent of pulmonary parenchymal involvement [38].
The FVC/DLCO ratio is the percentage predicted FVC/DLCO ratio (FVC%/DLCO%) and has been used as a predictor of the presence of pulmonary hypertension in the setting of ILD and scleroderma-related pulmonary hypertension [39, 40]. The principle behind the FVC%/DLCO% ratio is that in ILD both the FVC and DLCO fall, and their fall is proportional, whereas in pulmonary arterial hypertension, the DLCO falls significantly and out of proportion to the FVC. Several different threshold values for FVC%/DLCO% have been published, ranging from 1.4 to 2.2. It has been proposed that subjects with ILD with an FVC%/DLCO% ratio above the threshold are more likely to have pulmonary hypertension. However, the utility of the FVC%/DLCO% decreases with increasing emphysema, as seen in CPFE [41].
Further studies are required to assess the relevance of different spirometric parameters in the routine evaluation of this group of patients.
The role of echocardiography
Although echocardiography represents an important screening tool for detecting PH-ILD, confirmation with RHC is required. A risk of inaccuracy and/or overestimation in this group of patients has been reported more frequently in female subjects, in the presence of alterations of cardiac electrical activity, in cases of blood pressure values ≥140/90 mmHg and in patients being treated with furosemide [42].
While there are operator-dependent variabilities with the test, echocardiography provides several indicators of pulmonary hypertension and right heart function. The utility of echocardiography can be limited in patients with parenchymal lung disease because suboptimal images are often encountered. Good correlation has been shown between Doppler-assessed tricuspid regurgitant peak flow velocity (TRV) and haemodynamic parameters collected by means of RHC [43]. Likewise, a significantly increased sPAP is sensitive and specific to the presence of pulmonary hypertension [44, 45]. Unfortunately, both sPAP and TRV can be measured only in the presence of detectable tricuspid regurgitation. While this finding can be easily observed in severe pulmonary hypertension, it may pose limits in mild–moderate pulmonary hypertension. It has been recognised that even when conditions are optimal, echocardiographic estimates for right atrial and pulmonary artery pressures can differ significantly from those determined by RHC, with a trend to both overestimate and underestimate the pulmonary artery systolic pressure [46].
The sensitivity and accuracy of echocardiography compared to RHC has been investigated recently by Sonaglioni et al. [42] in a large cohort of patients with clinical suspicion of pulmonary hypertension. A moderate correlation was observed between sPAP and mPAP when evaluated by echocardiography and RHC (r=0.65 and r=0.60, respectively). A bias of −11.9 mmHg (with the 95% limits of agreement ranging from −45.4 to 121.5 mmHg) for sPAP evaluation and −4.6 mmHg (95% limits of agreement ranging from −45.4 to 121.5 mmHg) for mPAP estimation was observed in the Bland–Altman analysis, suggesting a general overestimation of PAP in the echocardiographic measurement [42]. Similar findings were reported by Arcasoy et al. [43] in their analysis of a cohort of patients affected by different severe lung diseases who were referred for lung transplantation. In a study by Keir et al. [47], the predictive utility of the updated European Society of Cardiology (ESC)/European Respiratory Society (ERS) transthoracic echocardiographic screening recommendations versus RHC was evaluated in a large group of well-characterised ILD subjects. ESC/ERS recommend tricuspid regurgitation velocity thresholds for assigning high (>3.4 m·s−1), intermediate (2.9–3.4 m·s−1) and low (<2.8 m·s−1) probabilities of pulmonary hypertension. Surprisingly, 40% of subjects were misclassified as low probability of pulmonary hypertension when pulmonary hypertension was confirmed by RHC. The authors found that the TRV screening thresholds were associated with a high positive predictive value (86%) for confirming pulmonary hypertension, but were of limited predictive value in excluding pulmonary hypertension. The sensitivity and specificity (n=37, sensitivity 92.3%, specificity 81.8%) of echocardiography was improved when a combination of parameters was used including the right ventricular systolic pressure (RVSP) (>43 mmHg), right ventricular outflow tract diameter (>34 mm) and the tricuspid annular plane systolic excursion (TAPSE) ≤18 mm [48].
Features on echocardiography that increase sensitivity, and pre-test probability for confirming suspected pulmonary hypertension on right heart catheterisation may include a TRV that is >3 m·s−1, evidence of right atrial and/or right ventricular dilatation, systolic interventricular septal flattening and any evidence of right ventricular dysfunction. This could include a reduced TAPSE, or a reduced peak lateral tricuspid annular systolic velocity.
Circulating biomarkers, such as brain natriuretic peptide (BNP) or N-terminal (NT)-proBNP, are useful tools for screening patients with suspected pulmonary hypertension. Although BNP or NT-proBNP values have less sensitivity and specificity in moderate pulmonary hypertension, they could be higher in severe PH-ILD [49, 50]. The importance of BNP measurement in ILD has been demonstrated recently, not only for diagnostic screening, but also for risk evaluation. A combination of BNP level and RVSP determined by echocardiography provided a better prediction of mortality than the RVSP alone [51]. When BNP is combined with DLCO and findings on echocardiogram, it provides a good indicator of the presence of pulmonary hypertension and provides an opportunity to risk-stratify patients [52].
The role of chest computed tomography
Several features seen on chest computed tomography can raise suspicion for PH-ILD. These include right ventricular enlargement, and several measures related to pulmonary artery enlargement. Approaches to assessing pulmonary artery enlargement include the diameter of the pulmonary artery before bifurcation of the right and left pulmonary arteries, and the pulmonary artery (PA)/aorta (A) ratio >1; in fact, patients with a PA/A >1 have a higher risk of death or transplant [53]. Right ventricular enlargement can be assessed using the right ventricle/left ventricle ratio (>1) and flattening of the interventricular septum. A pulmonary artery diameter of >25 mm in patients with ILD had a sensitivity of 86.4%, but a specificity of only 41.2% [54].
The role of exercise testing
IPF patients may develop a progressive exercise limitation during the course of the disease. The 6-min walk test (6MWT) can be useful for defining changes in exercise tolerance, because it is easy to perform, reproducible and correlates with daily activity performance. Typically, during the 6MWT, the distance covered during the test (6MWD), oxygen saturation, heart rate and Borg dyspnoea scale are recorded.
Although it has been widely used in studies investigating new treatments for PAH, 6MWT outcomes in ILD patients can be more difficult because any limitation reflects both the vascular component and parenchymal involvement. Furthermore, the causes underlying the exercise limitation cannot be extrapolated from data collected by the test.
The negative correlation between 6MWD and mortality in ILD is controversial [9, 55–62]; however, oxygen saturation during the test, slow heart rate recovery during the first minute of rest and alterations in chronotropic response appear to provide prognostic insight regarding mortality in this group of patients. For all these reasons, oxygen and heart rate measurements obtained during the 6MWT should be considered in ILD patients.
Progressive pulmonary vascular remodelling in the setting of ILD results in a reduced pulmonary capillary bed and impaired pulmonary arterial recruitment giving rise to pathophysiological abnormalities that are enhanced with exercise. Abnormalities can result from reduced pulmonary blood flow and cardiac output, worsening ventilation perfusion mismatching, and progressive ventilatory inefficiency. These abnormalities lead to progressive worsening of aerobic capacity and exercise tolerance. Cardiopulmonary exercise testing (CPET) can augment the pathophysiological abnormalities that pulmonary hypertension imposes on patients with ILD [63]. These pathophysiological derangements are responsible for characteristic differences observed during CPET and potentially identify patients who are at increased risk of pulmonary hypertension. Despite all these observations, a noninvasive test providing high sensitivity and high specificity in detecting PH-ILD has not yet been identified, and RHC remains the gold standard for pulmonary hypertension assessment, even in this group of patients.
Haemodynamic assessment
RHC is not routinely performed in patients affected by chronic lung disease; however, according to the 6th WSPH recommendations, it should be performed when the presence of pulmonary hypertension can influence management of the underlying lung disease (such as referral for transplantation, inclusion in clinical trials or registries, treatment of unmasked left heart dysfunction or the use of compassionate treatments) [64]. In addition, data obtained by RHC may provide insight into the setting of progressive exercise limitations and/or gas exchange abnormalities not explained by ventilatory impairment or when a precise prognostic evaluation is required.
In chronic lung disease, significant changes in intrathoracic pressure may occur during respiratory cycles, and the measurement of RHC values must be very accurate. In fact, increases in intrathoracic pressure and changes in end-expiratory lung volumes due to expiratory muscle contraction and dynamic hyperinflation may affect pulmonary pressure during the test. To avoid these biases, it is recommended to measure average pulmonary artery pressure over several respiratory cycles, particularly in the measurement of pulmonary capillary wedge pressure which is subject to wide variations in pleural pressure [65].
Prognosis in PH-ILD
As stated, the presence of pulmonary hypertension in the setting of ILD portends a poor prognosis [9]. IPF patients with associated pulmonary hypertension exhibited a higher 1-year mortality rate compared to a matched cohort of candidates for lung transplantation without pulmonary hypertension. Similar findings have encouraged referral for lung transplantation without delay [66]. The presence of pulmonary hypertension (mPAP ≥25 mmHg) has been found to be an independent predictor of mortality in a large cohort of IPF patients referred for lung transplantation [11]. In fact, high mPAP values measured at the time of the initial evaluation of IPF patients with RHC are an independent predictor of survival using multivariate analysis [67]. Interestingly, in the same study, Kimura et al. [67] observed a very similar prognosis in patients with mPAP values of 21–25 mmHg and those with mPAP ≥25 mmHg. Based on reduced oxygen concentration analysis, an mPAP value of 20 mmHg appeared to be a reasonable cut-off for predicting prognosis and mortality [67].
While mPAP and sPAP are often the focus in the assessment of pulmonary hypertension, PVR has also been demonstrated to provide prognostic data. In patients with severe ILD who underwent RHC, PVR was reported to be a strong predictor of 1-year mortality independently of a specific diagnosis of IPF. Moreover, PVR was a stronger predictor of mortality compared to mPAP [68]. Recently, as observed in data from the COMPERA registry, the presence of PVR greater than ∼5 Wood Units was associated with a worse survival compared to a PVR ≤5 Wood Units [69]. In patients with advanced ILD, a complete haemodynamic assessment can provide important prognostic information, driving the management of this clinically difficult population.
Pharmacological management of PH-ILD
Before considering any pulmonary vasodilator therapy, treatment directed at the underlying lung disease should be optimised. This could include antifibrotic and immunosuppressive therapy. Given the significant impact of PH-ILD, eligible patients should be referred for lung transplantation evaluation. In patients with associated hypoxic respiratory failure, long-term oxygen treatment is intuitively useful, although it has been clearly evaluated only in COPD. In patients with COPD, the use of oxygen for ≥15 h·day−1 was useful in preventing the progressive increase of mPAP [66, 70], and if used for ≥18 h·day−1, a slight decline in mPAP was observed [70, 71]. However, in COPD patients with moderate desaturation at rest or during exercise, the use of oxygen does not improve survival or hospitalisation [72]. The use of supplemental oxygen in ILD has been inferred from data from patients with COPD. Studies to date have been inconclusive as to the effect of oxygen effect on exercise capacity, quality of life, survival or dyspnoea in patients with ILD [70, 73–75].
Until recently, most clinical trials of pulmonary vasodilators in PH-ILD have been disappointing (table 1). There are concerns that some of the negative findings could be more related to trial design than true lack of efficacy of the drug being studied. Most PH-ILD trials enrolled patients with pulmonary hypertension diagnosed only based on diffusion capacity and/or echocardiography criteria. They did not require RHC-derived haemodynamic parameters, raising concerns that the wrong population was enrolled in the study. Few of the previous trials have focused specifically on severe PH-ILD. Another important consideration is that PH-ILD trials are difficult to complete because of the impact of dual disease states and the rarity and short life expectancy of patients. Another limitation is hesitation among clinicians to enrol patients with advanced disease for placebo-controlled trials.
Randomised controlled trials (RCTs) of pulmonary vasodilators in pulmonary hypertension (PH)-associated interstitial lung diseases (ILDs)
Previously, there was considerable interest in the use of endothelin receptor antagonists (ERAs) in ILD, as these medications are believed to have antifibrotic properties in addition to their known effects as pulmonary vasodilators. King et al. [76] compared bosentan to placebo in a RCT of 158 patients with PH-ILD that failed to demonstrate benefit in any of the parameters studied, including 6MWT distance and time to clinical worsening or death. A subsequent larger trial also failed to show any benefit with bosentan [79]. In a study of macitentan in pulmonary hypertension that included patients with ILD and COPD, no benefit was seen. Rates of discontinuation due to an adverse event were higher in ILD and COPD patients than in non-ILD/COPD including PAH patients. A higher hospitalisation rate and worse survival were observed in COPD patients at 12 months compared to the other subgroups [88]. Given the consistently demonstrated lack of efficacy and potential harm demonstrated in these studies, the use of ERAs is not suggested in PH-ILD.
ARTEMIS-PH examined the effects of ambrisentan in patients with IPF with RHC-confirmed pulmonary hypertension. This study was terminated early after a subgroup analysis of patients with pulmonary hypertension in the ARTEMIS-IPF study failed to suggest benefit. In fact, at the interim analysis, a significantly higher rate of lung function decline and an increase in the rate of hospitalisation related to respiratory impairment was observed in the treatment arm [50]. The RISE-IIP study of riociguat, a soluble guanylate cyclase stimulator approved for patients with both group 1 and group 4 pulmonary hypertension, in patients with IIPS with RHC-confirmed pulmonary hypertension was terminated early for increased rates of serious adverse events and death in the treatment group. The study failed to demonstrate improvement in 6MWD, or time to clinical worsening events in patients treated with riociguat [81]. In the STEP-IPF trial [78], patients with advanced IPF enriched for pulmonary hypertension by means of reduced DLCO failed to demonstrate any difference in the primary end-point of a ≥20% increase in 6MWD. However, there was a trend toward a mortality benefit at 24 weeks in the treatment arm (p=0.07). A pre-specified post hoc analysis of those with echocardiographic data demonstrated that in patients with right ventricular dysfunction, sildenafil resulted in in better preservation of exercise capacity as compared with placebo. Sildenafil also improves quality of life in subjects with right ventricular hypertrophy and right ventricular dysfunction [89]. While several secondary end-points, including quality of life, arterial oxygen saturation and DLCO showed improvement, the study's primary end-point did not. Thus, the data from that trial suggest that sildenafil is unlikely to be harmful in this group of patients, but results should be interpreted with caution and further investigation is required to confirm the beneficial effects of sildenafil in this subgroup of patients.
A clinical trial investigated the use of sildenafil in severe IPF patients already treated with nintedanib, but there were no significant improvements in the symptoms seen. A subanalysis performed in patients with or without echocardiographic signs of right heart dysfunction did not show any subjective differences or changes in FVC with nintedanib plus sildenafil versus nintedanib alone [90].
In addition, similar findings were seen in a phase IIb study investigating treatment with sildenafil and pirfenidone in patients with advanced IPF and an intermediate or high probability of group 3 pulmonary hypertension [91]. Thus far, the use of these drugs should be considered only in expert centres for selected patients or in clinical trials, and they require careful evaluation.
Recently, the INCREASE trial assessed the safety and efficacy of inhaled treprostinil versus placebo in patients affected by PH-ILD, using the change in 6MWD as the primary end-point [87]. After 17 weeks of treatment, the walk distance increased by 21.1 m in the treatment arm, while it decreased by 10 m in the placebo group. The study met its primary efficacy end-point of change in 6MWD and all secondary end-points. There were significantly fewer clinical worsening events, and a decrease in underlying disease exacerbations in patients receiving inhaled treprostinil compared to placebo. Improvements in the safety end-point, which was FVC, were also observed in the treatment group. The findings from INCREASE may suggest a role for inhaled treprostinil in improving disease outcomes and preventing progression [87]. A subgroup analysis of patients with IPF demonstrated that treatment with inhaled treprostinil was associated with significant improvements in FVC. Improvements of 84.52 mL and 168.52 mL (n=92, p=0.0108) at weeks 8 and 16, respectively, and improvements in the FVC at weeks 8 (2.54% predicted; p=0.0380) and 16 (3.50% pred; p=0.0147) were seen compared to placebo [92]. This change was greater in IPF patients than in connective tissue disease-associated ILD and CPFE patients, leading to the hypothesis that inhaled treprostinil might reduce fibrosis progression, contributing to the improvement in 6MWD [92, 93].
The mechanism by which treatment with treprostinil to an increase in FVC is unclear. It will be important to investigate whether this finding is related to a change in pulmonary vessel compliance or due to a direct antifibrotic effect of treprostinil as observed in vitro and in mice [94, 95]. The TETON trial is currently underway, investigating the safety and efficacy of inhaled treprostinil in IPF patients, using the absolute FVC as the primary end-point (clinicaltrials.gov identifier NCT04708782).
The use of systemic treatments approved for patients with PAH in group 3 pulmonary hypertension has always raised concerns due to the potential for worsening ventilation/perfusion (V′/Q′) matching. Interestingly, as observed in other studies, inhaled drugs preferentially drive blood flow to the lung zones with better ventilation, reducing the risk of a V′/Q′ mismatch [96, 97]. Clinical trials investigating the use of inhaled treprostinil are ongoing in patients affected by pulmonary hypertension related to sarcoidosis (clinicaltrials.gov identifier NCT03814317) and COPD (clinicaltrials.gov identifier NCT03496623).
Pulmonary rehabilitation
Patients affected by several chronic lung disorders, especially in advanced stages of the disease, complain of dyspnoea, fatigue, reduced exercise tolerance and skeletal muscle dysfunction. Exercise capacity and quality of life in COPD patients have been shown to improve with pulmonary rehabilitation, along with a reduction in symptom burden and hospitalisation rates [98–101].
In IPF patients, greater 6MWD improvement after pulmonary rehabilitation was associated with a higher FVC at baseline. These findings suggest that IPF patients realise greater benefits from pulmonary rehabilitation with milder disease, supporting the importance of early referral in this group of patients [102]. Pulmonary rehabilitation had a positive impact on functional status and quality of life in 402 consecutive ILD patients, the majority of whom had IPF, where 6MWD improved by mean±sem 46±3 m, and there was a slight increase in FVC. In addition, in a subgroup affected by pulmonary hypertension related to ILD, benefits were observed [103]. However, tailored protocols for each disease might be useful for taking into consideration all the different pathological mechanisms underlying each disease.
Conclusion
It is increasingly recognised that the presence of pulmonary hypertension in ILD is associated with a worse quality of life and a poor prognosis. Out of proportion or progressive changes on serial testing that includes pulmonary function testing including the DLCO, impairment in 6MWD or performance on cardiopulmonary exercise testing, and changes on echocardiography, could suggest the development of pulmonary hypertension in ILD patients. Because of the poor prognosis in PH-ILD, screening and appropriate and timely haemodynamic assessments are encouraged. Although transthoracic echocardiography is the most common tool used to investigate pulmonary hypertension in this group of patients, it lacks sensitivity and specificity, and RHC remains the gold standard for this diagnosis. The use of pulmonary vasodilators in these patients is still debated, and several clinical trials have been interrupted prematurely due to increases in mortality and hospitalisation in the treated arms. Recently, given the positive results of the INCREASE trial, which evaluated the safety and efficacy of inhaled treprostinil in PH-ILD, the opportunity for treatment of this group of patients should be considered.
Footnotes
Provenance: Commissioned article, peer reviewed.
Conflict of interest: A.B. Waxman reports grants from United Therapeutics, Gossamer and Altavant, and personal fees from ARIA-CV and Acceleron, outside the submitted work.
Conflict of interest: D. Elia has nothing to disclose.
Conflict of interest: Y. Adir reports personal fees from TEVA, SANOFI, Bayer and Pfizer, and grants and personal fees from GSK, AstraZeneca and Janssen, outside the submitted work;
Conflict of interest: M. Humbert reports grants or contracts from Acceleron, Janssen and Merck to his institution; consulting fees from Acceleron, Janssen and Merck (Steering Committees for pulmonary hypertension); payment or honoraria for lectures, presentations, speakers’ bureaus, manuscript writing or educational events from AOP, Janssen and Merck; and participation on Advisory Boards for Acceleron, Janssen, Merck and United Therapeutics.
Conflict of interest: S. Harari reports personal fees from Roche, grants and personal fees from AstraZeneca and Boehringer Ingelheim, outside the submitted work.
- Received October 4, 2021.
- Accepted May 23, 2022.
- Copyright ©The authors 2022
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References











Jump To
- Article
- Abstract
- Abstract
- Introduction
- Aetiology and classification
- Haemodynamic classification
- Screening and diagnostic evaluation
- The role of pulmonary function testing
- The role of echocardiography
- The role of chest computed tomography
- The role of exercise testing
- Haemodynamic assessment
- Prognosis in PH-ILD
- Pharmacological management of PH-ILD
- Pulmonary rehabilitation
- Conclusion
- Footnotes
- References
- Figures & Data
- Info & Metrics