





Abstract
COPD is predicted to become the third leading cause of morbidity and mortality worldwide by 2030. Cigarette smoking (active or passive) is one of its chief causes, with about 20% of cigarette smokers developing COPD from cigarette smoke (CS)-induced irreversible damage and sustained inflammation of the airway epithelium. Inflammasome activation leads to the cleavage of pro-interleukin (IL)-1β and pro-IL-18, along with the release of pro-inflammatory cytokines via gasdermin D N-terminal fragment membrane pores, which further triggers acute phase pro-inflammatory responses and concurrent pyroptosis. There is currently intense interest in the role of nucleotide-binding oligomerisation domain-like receptor family, pyrin domain containing protein-3 inflammasomes in chronic inflammatory lung diseases such as COPD and their potential for therapeutic targeting. Phytochemicals including polyphenols and flavonoids have phyto-medicinal benefits in CS-COPD. Here, we review published articles from the last decade regarding the known associations between inflammasome-mediated responses and ameliorations in pre-clinical manifestations of CS-COPD via polyphenol and flavonoid treatment, with a focus on the underlying mechanistic insights. This article will potentially assist the development of drugs for the prevention and therapy of COPD, particularly in cigarette smokers.
Abstract
This review compiles current investigations into the role of polyphenols/flavonoids in the alleviation of cigarette smoke-induced inflammasome; notably it provides a promising hit for rectifying the treatment of COPD. https://bit.ly/36OcUO9
Introduction
COPD has an increasing prevalence and is anticipated to become the third main cause of morbidity and mortality in the global population by 2030 [1]. Vitally, COPD can be caused by prolonged exposure to harmful particles, particulates and gases, infection with acute or chronic inflammatory conditions, or by injuries that include the airways, pulmonary vasculature and lung parenchyma. COPD is categorised by persistent airflow obstruction instigated by exposure to irritants; for instance, cigarette smoke (CS), dust and fumes [2]. Pulmonary injury comprises the phases of initiation (by exposure to CS, pollutants, infectious pathogens and agents), progression and consolidation. The involvement of complex interactions among oxidative stress, inflammation, extracellular matrix proteolysis, autophagic and apoptotic cell death leads to tissue damage [3]. The inflammatory response to progressive inflammation and tissue oxidative stresses leads to airway remodelling, which limits airflow and causes airway obstructions and tissue destruction, resulting in a decrease of forced expiratory volume [4]. In other airway obstructions, a major feature of COPD is pulmonary emphysema, which is described as structural changes and destruction of the alveolar air sacs that lead to decreased gas-exchanging surfaces and impaired gas exchange [5]. From a physio–pathological viewpoint, COPD is concomitant with chronic inflammation influencing the lung parenchyma and peripheral airways that cause irreversible and progressive airflow limitations. This inflammation is indicated by augmented numbers of alveolar macrophages, neutrophils, T-lymphocytes (cytotoxic T-cell type 1, T-helper cell (Th) 1 and Th17 cells) and intrinsic lymphoid cells engaged from the circulation [6]. These cells, as well as structural cells encompassing epithelial and endothelial cells and fibroblasts, secrete a variety of pro-inflammatory mediators, including cytokines, chemokines, growth factors and lipid mediators [7]. Interleukin (IL)-18 is a pro-inflammatory cytokine that was first described as an interferon (IFN)-γ-inducing factor. Similar to IL-1β, IL-18 is synthesised as an inactive precursor that requires processing by caspase-1 into an active-form cytokine [8]. Moreover, several studies used a mouse model to show that IL-18 over-expression results in emphysematous lesions. The data prompts the hypothesis that IL-18 induces an extensive range of COPD-like inflammatory and remodelling responses in the murine lung and induces mixed IL-1, IL-2 and IL-17 responses. IL-18 has been identified as a latent target for future COPD therapeutics to restrain the damaging and remodelling processes arising in COPD lungs [9].
Inflammasome
The functions of innate immune responses act as a first-line defence upon exposure to potentially deleterious stimuli. The innate immune system has evolved various extracellular and intracellular receptors that carry out surveillance for possibly detrimental particulates. Inflammasomes are defined as intracellular innate immune multiplex proteins that are formed and activated upon subsequent interaction with these stimuli [7]. The canonical inflammasomes can be elicited by pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) [10]. Two families of pattern-recognition receptors (PRRs), including the nucleotide-binding domain, leucine-rich repeat containing proteins (NLRs) and absent in melanoma 2 (AIM2)-like receptors, act as sensor components to stimulate the inflammasome [11]. The NLR family consists of several subclasses, including nucleotide-binding oligomerisation domain-like receptor family, pyrin domain containing protein (NLRP) 1, NLRP3, NLRP6, NLRP12 and NLR family CARD domain-containing protein 4 (NLRC4; also known as interleukin converting enzyme protease activating factor (IPAF)). In fact, the NLR family has two common features: the first one is a nucleotide-binding oligomerisation domain, which is bounded by ribonucleotide-phosphates, and the second one is the C-terminal leucine-rich repeat (LRR), which functions as a ligand-recognition domain [12]. In addition, the AIM2-like receptor (ALR) family encompasses two subclasses: AIM2 and interferon gamma inducible protein (IFI) 16. The common feature of the ALR family is PRRs with one or two hematopoietic, IFN-inducible and nuclear (HIN) domains at the C-terminal and a pyrin domain (PYD) at the N-terminal that can bind with cytoplasmic double-strand DNA (dsDNA) to initiate the inflammasome pathway [13].
In mammals, four major types of inflammasome, including NLRP1, NLRP3, NLRC4 and AIM2, have been confirmed by numerous studies of the signalling mechanisms and their functional effects. The activation of different inflammasomes may contribute to important immune response functions or cause cell pyroptosis [14]. The major domains of different inflammasome protein components can be classified as: caspase recruitment domain (CARD), domain with function to find (FIIND), HIN-200/IF120x domain (HIN), LRR, NAIP (neuronal apoptosis inhibitory protein), CIITA (MHC class II transcription activator), HET-E (incompatibility locus protein from Podospora anserina) and TP1 (telomerase-associated protein) (NACHT), and PYD. In general, an unoligomerised inflammasome may consist of two or three protein subunits. The unoligomerised NLRP1 inflammasome is composed of NLRP1 and caspase-1 (CASP1) protein subunits [15]; the unoligomerised NLRP3 inflammasome is composed of NLRP3, ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain) and CASP1 protein subunits [16]; the unoligomerised IPAF (NLRC4) inflammasome is composed of IPAF and CASP1 protein subunits [17]; and the unoligomerised AIM2 inflammasome is composed of AIM2 and CASP1 protein subunits [18]. The NLRP1 protein comprises five domains: PYD, NACHT, LRR, FIIND and CARD; the NLRP3 protein encompasses three domains: PYD, NACHT and LRR; the ASC protein includes two domains: PYD and CARD; the IPAF protein contains three domains: CARD, NACHT and LRR; the CASP1 protein consists of three domains: CARD, p20 and p10. All four major inflammasomes possess one unique CASP1 protein subunit, and the CASP1 can link to the other inflammasome components via CARD–CARD interaction. The activation on PRRs of inflammasome may cause the autocleavage of CASP1 to release the active-form caspase-1 [10]. Inflammasomes are the intracellular multiprotein complexes that can directly or indirectly trigger one or more caspase signalling pathways to process and secrete various pro-inflammatory cytokines, including IL-1α, IL-1β, IL-18 or transforming growth factor-β (TGF-β), and subsequently engage innate immune responses [19, 20].
During bacterial infection, NLRP1 may be responsible for pathogen recognition and resistance. The NLRP1 inflammasome pathway could be activated by various stimuli, i.e. pathogen lethal toxin, muramyl dipeptide or a reduction in intracellular ATP level [15]. Upon cytoplasmic infection by Gram-negative bacteria, NAIPs act as upstream sensors that bind with bacterial ligands and then co-assemble with NLRC4 to constitute NLRC4 inflammasome, which leads to the release of active-form caspase-1 [21, 22]. Of note, AIM2 inflammasome, a cytosolic dsDNA-sensing inflammasome, takes part in host defences and immune responses to DNA virus infections as well as intracellular bacterial infections [23, 24]. During viral infection, mitochondrial misfunction or loss of membranous integrity, dsDNA can appear in the cytoplasm, and that may elicit AIM2 inflammasome pathways [25]. Some investigations have revealed the involvement of AIM2 inflammasome expression in subsequent accelerated COPD development. The redistribution of AIM2 from nucleus to cytoplasm and co-localisation with the cleaved IL-1β indicates that AIM2 inflammasome can be prompted in the airway epithelium and macrophages of COPD patients and CS-exposure mice [26]. An in vitro study showed that the expression of AIM2 in COPD-derived CD14+ peripheral blood mononuclear cells is higher than that in nonsmokers and smokers [26, 27]. The activation of AIM2 inflammasome in a caspase-1- and caspase-4-dependent fashion can cause IL-1α release but not IL-1β, and that can further promote TGF-β release as pro-fibrotic processes [28]. An in vivo study exhibited that the recruited macrophages or dendritic cells in smoking mice increased the AIM2 expression associated with caspase-11 (analogue with human caspase-4) but not caspase-1. CS exposure still caused an induction of alveoli enlargement in caspase-11 dysfunctional (knockout) mice, but the deposition of collagen in the bronchioles was lower than that in CS-exposure normal mice [27]. NLRP3-knockout mice have protective effects on the CS-induced COPD model, whereas IPAF- or AIM2-knockout mice have no protected potential in the same CS-challenge COPD model [29]. According to the limited available evidence, the pathologic mechanisms of AIM2 inflammasome in CS-induced COPD are different from the NLRP3 inflammasome pathway, and it are permeated by caspase-4 and TGF-β release to promote bronchial tissue fibrosis and exacerbated COPD.
In all inflammasomes pathways, the NLRP3 inflammasome could be activated in response to the various types of stimuli by PAMPs and DAMPs, such as toxins, extracellular ATP, cytokines, ion fluxes, endoplasmic reticulum (ER) stress and oxidative stress [30, 31]. The NLRP3 inflammasome entails a cytosolic PRR, an adaptor molecule ASC and the protease precursor procaspase-1. Assembly of the NLRP3 inflammasome complex causes the cleavage and activation of caspase-1 that triggers processing, which releases IL-1β and IL-18 cytokines and causes cell death via pyroptosis [32]. Some inducers, i.e. lipopolysaccharide (LPS), act on the canonical NLRP3 inflammasome pathways to increase cytoplasmic active-form caspase-1, and the synchronous trigger of noncanonical inflammasome pathways to activate caspase-4/11, caspase-5 or caspase-8 as well, and that may modulate cell death by apoptosis or necrosis [33–35].
Macrophages sense danger signals such as bacterial toxins or extracellular ATP derived from tissue damage or infection and initiate the activation of an intracellular molecular complex inflammasome. Pyroptosis is an inflammatory form of programmed cell death (PCD) that is found to be involved in the development of chronic inflammation, i.e. COPD, which is activated by the inflammatory caspase cleavage of gasdermin D (GSDMD) and apoptotic caspase cleavage of gasdermin E [20, 23]. It is critical that the activation of the inflammatory response and inflammasome are tightly regulated [36]. In the immunopathology of COPD, immune and nonimmune inflammatory alterations with oxidative stress imbalances are found, as well as changes in the protease/anti-protease ratio caused by direct and indirect genetic and epigenetic-environmental defects. COPD leads to irreversible tissue damage and chronic inflammation with aberrant tissue repairs, which induces chronic obstruction of the airway, bronchitis and systemic damage [2, 9]. Evidence from a previous review suggested the role of inflammasomes in the pathogenesis of numerous chronic respiratory diseases and acute lung injuries, such as asthma, transfusion-related acute lung injury (ALI), ventilator-induced lung injury, COPD and pulmonary fibrosis [37].
From a physiological viewpoint, the respiratory system provides gas exchange from the external environment for metabolism. The airway and lung are incessantly exposed and challenged to a variety of inhaled pathogen agents, harmful particulates and internal self-derived dangerous signals during their whole lifespan. The innate immune reaction plays a vital role in protecting the pulmonary system from infection and disease, and inflammasome pathways act on the initiation of innate immune responses. Continuously and seriously activated inflammasomes in epithelial cells, macrophages or other immune cells may cause an increasing release of several pro-inflammatory cytokines and the occurrence of cell pyroptosis, which subsequently result in pulmonary tissue remodelling, fibrosis and COPD. Thereby, this article comprehensively reviews and discusses the effects of polyphenols and flavonoids on the inflammasome pathways in CS-induced COPD to provide a deep insight into the benefits of natural medicinal products as pharmaceutics.
Polyphenols and flavonoids
Plants that harbour secondary metabolites such as alkaloids, polyphenols, flavonoids, terpenoids and other specific natural compounds have gained a great deal of attention for the treatment of several clinical complications. Considering the promising results obtained using medicinal plants, with few/no side effects, as well as their easy attainment, comprehensive research on herbal plants to treat disease should be anticipated [38–40]. Polyphenols are natural composites found in many plants. Mostly, the polyphenols are categorised in three major classes: flavonoids, lignans and nonflavonoids (stilbenes and phenolic acids). Flavonoids comprise a large class of food components encompassing the flavone, isoflavanone, flavanone, flavonols catechin and anthocyanin subclasses [41–43]. Remarkably, several biological properties of polyphenols have been scientifically demonstrated, including antiallergic, antiviral, antibacterial, anticarcinogenic, anti-inflammatory, antithrombotic, vasodilatory and hepatoprotective effects [44, 45], as well as benefits in age-associated diseases including cancer, diabetes, Alzheimer's, osteoporosis and Parkinson's [46].
The pathophysiology of CS-COPD
With cigarette smoking (active or passive) being one of the chief causes of its occurrence, about 20% of cigarette smokers develop COPD. Furthermore, estimates suggest that from 25% to 33% of COPD patients are nonsmokers. Notably, CS exposure has been observed to result in irreversible damage and sustained inflammation of the airway epithelium, which might lead to COPD, although the exact pathophysiology remains elusive. CS induces the gathering of inflammatory cells (macrophages, neutrophils and lymphocytes), cytokine production, the triggering of inflammasome components (NLRP3, ASC, caspase-1), depression-related behaviours, and the enhancement of glucocorticoid receptor (GR) signalling. One in vivo study investigated the relationship and underlying molecular mechanism of CS-exposure, COPD and depression, and the evidence indicated that glucocorticoid resistance was manifested during central nervous system inflammation due to CS exposure, and a potential crosstalk-underlined mechanism between the brain and lung was found [47].
Recent studies have revealed that mitochondria are engaged in innate immune signalling that shows critical features in CS-induced inflammasome activation, pulmonary inflammation and tissue-remodelling responses. Mitochondrial dysfunction might increase intracellular oxidative stresses, decrease intracellular ATP level and cause mitochondrial DNA leakage into cytoplasm to trigger different inflammasome pathways. It has been revealed that mitochondria play a crucial role on the pathogenesis of CS-induced COPD [48]. While earlier studies have explored the character of membrane-bound Toll-like receptors in CS-induced inflammation, scarce information is available about the role of cytosolic NLRs in modulating CS-mediated inflammatory responses. The key role of the P2X7–NACHT, LRR and PYD domain-containing protein 3–ASC–caspase-1/11–IL-1β/IL-18 axis in CS-induced airway inflammation highlights this pathway as a probable therapeutic target for the treatment of COPD [49]. Triggering receptors expressed on myeloid cells 1 (TREM-1) is a crucial signalling receptor that can amplify pro-inflammatory innate immune responses. The activation of TREM-1 can aggravate the formation of inflammation by activating NLRP3 inflammasome pathways, and blocking TREM-1 may inhibit oxidative stress and decrease pro-inflammatory cytokine expression in order to alleviate ALI [50]. There are two proteins involved in the intercellular communication of epithelial cells: connexins 40 and 43 (Cx40, Cx43). Several studies have indicated that oxidative stress, described as the overproduction of 4-hydroxy-2-nonenal (4-HNE), is a crucial factor in pulmonary fibrosis [51]. CS exposure might induce the activation of nuclear factor-κB (NF-κB) and increase the formation of 4HNE-Cx40 adducts that may cause lung epithelium losses to intercellular junctions and remodelling.
After exposure to CS extract (CSE), 16 human airway epithelial (16HBE) cells increase lactate dehydrogenase release, upregulate the transcription and translation of NLRP3 inflammasome, and augment caspase-1 activity; enhanced IL-1β and IL-18 cytokine release has also been observed. In addition, NLRP3 is required to activate caspase-1. The results suggest that 16HBE cells treated with NLRP3 small interfering RNA or a caspase-1 inhibitor to silence NLRP3 expression can cause a decrease of IL-1β release and cell pyroptosis. CSE-induced inflammation contributed to pyroptosis via the reactive oxygen species (ROS)–NLRP3–caspase-1 pathway in the 16HBE cells. Thereby, NLRP3 inflammasome participated in CSE-induced cell damage and pyroptosis, which affords a new insight into COPD [52].
In general, the activation of NLRP3 inflammasome and subsequent cytokine secretion is a life-threatening step for innate immune responses. The activation of NLRP3 inflammasome is triggered by several internal and external factors and consequently results in inflammatory cytokine secretion. Inflammasome formation and activity play risky roles in several pathologies of diseases, such as cardiovascular, digestive, lung, metabolic, renal and central nervous system diseases [53]. CS and CSE exposure may induce inflammation and contribute to epithelial cell pyroptosis through the ROS–NLRP3–caspase-1 pathway in airway and lung tissues [52]. CS exposure leads to acute and chronic injury in lung tissues, as well as acting on the endothelial cells of the cardiovascular system to cause cell pyroptosis and the shrinkage of tunica intima/endocardium and epithelium in the bladder. The activation of NLRP3 inflammasome in endothelial cells by inducible nitric oxide synthase (iNOS) following CS exposure may be mediated by the soluble guanylyl cyclase–cyclic guanosine monophosphate–protein kinase G–tumour necrosis factor-α (TNF-α)-converting enzyme (TACE)–TNF-α pathway [54] as well as CS-induced pyroptosis of bladder tissues by activated the ROS–NLRP3–caspase-1 signalling pathway through the NLRP3 inflammasome activation relative pathways [55]. The nicotine in CS can decrease cell viability and induce ROS generation by triggering the NLRP6 inflammasome and provoke ER stress in human kidney cells to cause chronic kidney disease [56]. GSDMD undergoes proteolytic cleavage with caspase-1 to release its N-terminal fragment, which in turn mediates IL-18 and IL-1β secretions and causes pyroptosis. Pyroptosis is an inflammatory form of PCD, defined as being caspase-GSDMD-dependent. The NLRP3 inflammasome plays an indispensable role in mediating GSDMD activation. Asthma or infections may induce airway inflammation and cell pyroptosis via NLRP3 signalling pathways to cause capase-1 activation. Activated caspase-1 catalyses pro-IL-1β and pro-IL-18 to form IL-1β and IL-18; however, it cleaves GSDMD to form GSDMD N-terminal fragment (GSDMDNterm), which assembles into GSDMDNterm membrane pores and releases pro-inflammatory cytokines, IL-1β and IL-18, and subsequently causes pyroptosis [57, 58]. Current evidence has demonstrated that the NLRP3 inflammasome signalling axis could functionally mediate CS-induced inflammation, cytokine release, mucus production and pyroptosis in airway mucosa, which might be a critical mechanism associated with CS-induced airway remodelling. The involvement of signal pathways or targets in CS-induced lung injury or COPD from current investigations is summarised in table 1. Moreover, the signal pathways or targets of the inflammasomes activated in COPD are illustrated in figure 1.
Summary of the involvement of signal pathways or targets in cigarette smoke (CS)-induced lung injury or COPD from current investigations

The relationships between current investigations of the signal pathways or targets of inflammasomes involved in COPD. ASC: apoptosis-associated speck-like protein containing a caspase recruitment domain; CS: cigarette smoke; CSE: cigarette smoke extract; DEP: diesel exhaust particle; eATP: extracellular ATP; eHSP70: extracellular heat shock protein 70kDa; GSDMD: gasdermin D; IL: interleukin; LPS: lipopolysaccharide; MAPK: mitogen-activated protein kinase; NALP: NACHT, LRR and PYD domains-containing protein; NF-κB: nuclear factor-κB; NLRP3: nucleotide-binding oligomerisation domain-like receptor family, pyrin domain containing protein-3; TLR: Toll-like receptor; UFP: ultrafine particulate.
Polyphenols treatment via inflammasome in a cigarette smoking model
Currently, there is great interest in the role of inflammasomes in chronic inflammatory lung diseases and their targeting potential for therapy [7]. As alternative therapies, phytocompounds have been broadly used for destruction of inflammatory responses before. Selected phytochemicals have shown inhibitory properties on NLRP3 inflammasome activity in in vitro and in vivo tests [53]. Some flavonoids perform anti-inflammatory effects through the obstruction of NF-κB and NLRP3 inflammasome, suppression of pro-inflammatory cytokine IL-1β, IL-2, IL-6, TNF-α and IL-17A production, down-regulation of chemokines, and decrease of reactive nitrogen species and ROS. The most effective flavonoids for inflammation and modified immune responses are apigenin, quercetin and epigallocatechin-3-gallate (EGCG), although other compounds are still under investigation and cannot be omitted. The promising future of these compounds in different therapies has been discussed [72]. EGCG is the most plentiful catechin in green tea and provides protection against oxidative stress, lipid peroxidation and inflammatory responses caused by CS. Treatment with EGCG can decrease ROS production and suppress the lipid peroxidation induced by CSE and 4-HNE–protein adduct formation in airway epithelial cells. Further, it can inhibit CSE-induced NF-κB activity to decrease pro-inflammatory gene expression (cyclooxygenase-2 (COX-2), IL-6, IL-8, NADPH oxidase 4 (NOX4), iNOS, intercellular adhesion molecule 1, matrix metalloproteinase-9, cyclin-D1) via inhibition of the CSE-induced mitogen-activated protein kinase (MAPK) pathway [73, 74]. EGCG can restore superoxide dismutase (SOD) and catalase activities in the lungs and decrease CS-induced goblet cell hyperplasia and emphysema [75]. CS promotes mucus secretion and has a strongly effects the synthesis and secretion of MUC5AC [76]. Theaflavins are flavonoids extracted from black tea and are major antioxidants with protective effects. These effects are ascribed to the following major polyphenols: theaflavin, theaflavin-3-gallate, theaflavin-3′-gallate and theaflavin-3,3′-digallate [77]. Oral treatments with theaflavins showed the inhibition of epidermal growth factor receptor (EGFR) activation by CS and relieved airway mucous hypersecretion to reduce the levels of mucin MUC5AC [78]. Alternatively, treatment with theaflavin-3,3'-digallate significantly promoted the anti-oxidation capacity of lung tissues and attenuated CSE-induced emphysema and lung injury. Theaflavin-3,3'-digallate can decrease the generation of ROS and the expression levels of TNF-α, IL-1β and IL-6 of BEAS-2B (human bronchial epithelial) cells and inhibits necroptosis via the mediation of p38 MAPK–receptor-interacting serine–threonine-protein kinase 3 (RIPK3)–mixed lineage kinase domain-like pseudokinase (MLKL) signalling pathways. The pro-inflammatory cytokine IL-1 is catalysed by capase-1 from activated NLRP3 inflammasome. The protective effects of theaflavin-3,3'-digallate might inhibit NLRP3 pathways [79].
Flavonoids, e.g. (-)-epicatechin, increase tripartite motif-containing protein 25 and nuclear factor erythroid 2-related factor 2 (Nrf2) protein expression to promote ubiquitin-mediated Keap1 degradation and inhibit the activation of NLRP3 inflammasome by CS stimulation [80]. In the CS-induced airway inflammation of rats, baicalin treatment can modulate the histone deacetylase 2 (HDAC2)–NF-κB–plasminogen activator inhibitor type-1 signalling pathway to attenuate inflammation [81]. In the CS-induced COPD model, the TREM-1 level was increased, which activated the NLRP3 inflammasome pathway to aggravate tissue inflammation and lung injury [68]. TREM-1 is a crucial signalling receptor that can amplify pro-inflammatory innate immune responses. The activation of TREM-1 can aggravate the formation of inflammations by activating NLRP3 inflammasome pathways. Blocking TREM-1 may inhibit oxidative stress and decrease pro-inflammatory cytokine expression in response to ALI [50]. TREM-1, as the macrophage's cell surface receptor, is involved in the spread of the inflammatory response to bacterial infections or LPS challenges. Pre-treatment with quercetin, resveratrol and tea polyphenols can inhibit the secretion/shedding of soluble TREM-1 [82–84]. Quercetin is one of polyphenols that functions as an ROS scavenger and adenosine monophosphate-activated protein kinase (AMPK) activator. Peripheral blood mononuclear cells from COPD patients treated with quercetin can activate AMPK and promote the expression of Nrf2, which was found to reverse CSE-induced corticosteroid insensitivity [85]. Quercetin can protect and reduce the oxidative stress of macrophages from CSE exposure via reducing the levels of leukocytes, oxidative stress, histological pattern alterations of pulmonary parenchyma and lung function alterations [86]. In vitro CSE-induced results showed that several fatty acid esters of quercetin-3-O-glucoside had a protective potential against nicotine-induced cell death, membrane lipid peroxidation, and ameliorated the inflammation biomarkers COX-2 and prostaglandin E2 expression [87]. An in vitro study has shown that jaboticabin and 3,3'-dimethyellagic acid-4-O-sulphate are polyphenols originating from jaboticaba and exhibit anti-inflammatory activities induced by CSE [88]. Extracts of Tussilago farfara have a high polyphenol content and excellent antioxidant and good antimicrobic properties [89]. CS-induced lung inflammation can be attenuated by using ethanol extract from T. farfara flower buds through mediating the NLRP3 inflammasome, NF-κB and Nrf2 pathways [90]. N-Acetylcysteine has shown the potential to improve the immune state of COPD patients and a COPD animal model by downregulating pro-inflammatory and inflammatory cytokines, including IL-1β, IFN-γ, TNF-α and IL-18, through modulating and suppressing the NLRP3 inflammasome pathways of macrophages [91]. Magnesium isoglycyrrhizinate (MgIG), glycyrrhizic acid, is an anti-inflammatory agent originally used for treating hepatitis. Animals treated with MgIG experienced reduced inflammatory cell infiltration and accumulation in broncho-alveolar lavage. Decreased IL-6 and TNF-α production was revealed in the serum of CS/LPS-induced COPD rats through the inhibition of NLRP3 pathways [80]. For protection against epithelial injury by CS exposure, corilagin can reduce CS-induced intercellular junction breakdown on epitheliums by inhibiting 4HNE-Cx40 adduct formation and NF-κB activation to diminish CS-induced epithelial changes [92].
Gallic acid (GA) is a natural phenolic composite with high free radical scavenging potential to act as an antioxidant and anti-inflammatory agent [93]. GA treatment can restrain elastase-induced neutrophil infiltration and elevate myeloperoxidase (MPO) activity. In addition, production of the pro-inflammatory cytokines IL-6, TNF-α and IL-1β is suppressed by phosphorylation of p65NF-κB and IκBα with the down-regulation of IL-1β/TNF-α/keratinocyte chemoattractant/macrophage inflammatory protein-2/granulocyte colony stimulating factor gene expression. GA also suppressed the influx of neutrophils and macrophages induced by CS and dampened the expression of TNF-α/MIP-2/KC genes. This could significantly restore the levels of glutathione reductase and catalase, as well as reduce xanthine oxidase activity in lung tissues to attenuate and modulate the tissue oxidative stress and inflammation induced by CS [94, 95]. Exposure to particulate matter (PM) from air pollution decreases Nrf2 expression; GA can restore antioxidant status through activating the Nrf2 pathway to prevent and attenuate lung injury from PM exposure [96]. In CS-induced acute and chronic lung injuries, supplements with pomegranate juice can significantly protect lung tissue from injuries and decrease the expression of inflammatory mediators, apoptosis and oxidative stress [97]. In CSE and porcine pancreatic elastase (PPE)-induced animal models, spray instillations with salvianolic acid B significantly improved the bioavailability about 30∼100 fold over oral administration. The anti-emphysema effects of salvianolic acid B can reduce CSE-induced apoptosis and lipid peroxidation, elevate phosphor-signal transducer and activator of transcription 3 and vascular endothelial growth factor (VEGF) expression, and stimulate lung cell proliferation to enable the reversal of alveolar structural destruction in vivo [98]. CSE may cause inflammation and increase the tissue oxidative stress that leads to decreased elastin formation of fibroblasts by inhibiting the enzyme activity of lysyl oxidase [99]. The bioavailability of isorhapontigenin by oral administration is high and it has good anti-inflammatory effects and pharmacokinetic properties [100]. In vitro cells treated with isorhapontigenin can effectively reduce the levels of ROS and display cytokine-suppressing effects through the inhibited phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) pathway, which is insensitive to corticosteroids [101]. Silymarin has been reported to attenuate the chronic inflammation and oxidative stress in a CS-induced COPD model and the anti-inflammatory effect of silymarin may thoroughly suppress the activity of the extracellular signal-regulated kinases (ERK)/MAPK pathway [102, 103]. Murine orally received isoflavones led to reduced CS-induced pro-inflammatory cytokine gene expression, decreased inflammatory cells in the bronchoalveolar lavage fluid (BALF) and shrunk pulmonary emphysema [104]. Curcumin is a naturally existing polyphenolic compound present in the rhizome of Curcuma longa belonging to the family Zingiberaceae. The biological activity of curcumin to regulate the function of multiple signal pathways or transductions is linked with the attenuation of acute and chronic diseases. Numerous pre-clinical and clinical studies have demonstrated that curcumin modulated several mediators in cell signal transduction pathways including Akt, AMPK, activator protein 1, β-catenin, enhancer binding protein α, ERK5, p38/MAPK, mammalian target of rapamycin (mTOR), MyD-88, NLRP3 inflammasome, Notch-1, Nrf2, P21 (RAC1) activated kinase 1, PI3K, peroxisome proliferator-activated receptor γ (PPARγ), Rac1, Shh, STAT3, TGF-β, Toll-like receptor (TLR)-4 and Wnt [105–107]. The use of adjuvants such as piperine, curcumin nanoparticles, liposomal curcumin and the curcumin phospholipid complex showed the enhanced bioavailability and therapeutic potential [105]. In vitro, curcumin treatment can activate the PPARγ pathway and inhibit the NF-κB pathway to attenuate CSE-induced cell death and inflammatory cytokine expression. In vivo CS exposure can down-regulate the PPARγ+ cell number in lung tissues; while curcumin treatment can restore it and hinder TNF-α and IL-6 levels of serum to mitigate CS-induced inflammation [106]. Oral curcumin administration can increase the antioxidant gene expression in alveolar macrophage and lung tissue, such as oxygenase-1, glutamate-cysteine ligase and glutathione reductase, increase MPO activity, and decrease the neutrophils and macrophages in BALF to decrease intratracheal PPE- and CS-induced pulmonary inflammation and emphysema in mice [107]. Effective anti-inflammation corticosteroids are widely used in clinical therapies for asthma and COPD treatment. The suppression of pro-inflammatory gene expression by corticosteroids is based on the recruitment of the transcriptional co-repressor HDAC2 into an activated GR complex [108]. Chronic oxidative stress in COPD lungs is impeded HDAC2 activity [109], which blocks steroid efficacy or causes desensitisation [110]. Curcumin can restore and promote HDAC2 expression and activity, which may reverse the steroid insensitivity of asthma or CS-induced COPD [111]. In ALI induced by a serious infection, e.g. sepsis, curcumin treatment can hinder NF-κB, NLRP3 and pyroptosis-related protein expressions and increase sirtuin 1 (SIRT1) expression to demonstrate the protective effects of curcumin on septic ALI through up-regulation of SIRT1 to modulate the NLRP3 inflammasome pathway [112]. The major limiting factor of curcumin medical applications is its low bioavailability. However, there is a new treatment formulation using dry powder inhalation of curcumin for COPD that can provide good solubility and dissolution [113]. There are several classes of polyphenols present in apple juice, including flavanols, dihydrochalcones, flavan-3-ols and phenolic acids. Apple phenolics function as strong anti-inflammatory agents by shielding tissues from inflammatory injury [114]. Animals that received the oral administrate of apple polyphenols had significantly decreased oxidative stress and expression of pro-inflammatory factors in the lungs, reduced CS-induced accumulation of inflammatory cells in airways and lung, which might be partially reduced by inhibiting the P38/MAPK signalling pathway [115]. Increasing studies have illustrated the favourable therapeutic effects of resveratrol against lung diseases by preventing ageing, inflammation, oxidative stress, fibrosis and cancer in vitro and in vivo. Treatment with resveratrol can hinder inflammatory cytokine IL-1β and stimulated anti-inflammatory cytokine IL-8 release from alveolar macrophages in COPD [116]. Additionally, resveratrol treatment attenuates CSE-mediated glutathione (GSH) depletion in alveolar epithelial cells by promoting GSH synthesis and Nrf2 activity [117].
Corticosteroids are inefficient for reducing IL-8 and granulocyte-macrophage colony-stimulating factor (GM-CSF) release from alveolar macrophages, and inflammatory mediators released from the airways smooth the muscle cells of COPD patients. Resveratrol treatment can reduce the release of IL-8 and GM-CSF and restore VEGF release from human airway smooth muscle cells in COPD [118]. The combinative treatment of resveratrol and dexamethasone can significantly reduce all inflammatory parameters [119]. Resveratrol treatment can upregulate SIRT1 and proliferator-activated receptor-γ coactivator-1α (PGC-1α) expression to decrease the level of malondialdehyde and increase activity of SOD [71, 120]. In an in vivo CS-induced or LPS treatment COPD model, oxidative stress and inflammation caused injury to respiratory and cardiovascular systems. Treatment with resveratrol could reduce cardiac oxidative damage, prevent left ventricular remodelling and restore the expression of SIRT1 in the hearts of aged rats with emphysema [121] and may have a protective effect against CSE-induced apoptosis of human bronchial epithelial cells [122]. In an elastase-induced pulmonary emphysema COPD model, the expression of VEGFA, Nrf2, manganese SOD and airspace volume were significantly augmented in the lung tissues of the resveratrol treatment group [123]. In a combined LPS/CE-induced COPD model, resveratrol could significantly decrease the inflammatory cells in BALF and the level of inflammatory-IL-17, IL-6, IL-8, TNF-α and TGF-β cytokines in the lungs [124].
In CS exposure-induced chronic bronchitis, naringin treatment can significantly reduce the concentrations of IL-8, leukotriene B4 and TNF-α in BALF and improve SOD activity in lung tissues [125]. Naringin is a flavanone glycoside found in citrus fruits and grapefruit that is characterised by the following effects: antioxidation, anti-peroxidation and anti-inflammation [126]. Naringin treatment can significantly reduce ovalbumin-induced coughs and airway hyper-responsiveness and inhibit inflammatory cell infiltration and IL-4, IL-5 and IL-13 in BALF [127]. Naringin can restore the IL-10 level, prevent CS-induced neutrophil infiltration and decrease the activation of MPO and MMP-9 with suppressed inflammatory cytokine release, such as TNF-α and IL-8, in a CS-exposed animal model [128]. Liquiritin apioside is a flavonoid from Glycyrrhiza uralensis that can protect lung epithelial cells from CS-induced injury by increasing the levels of anti-oxidative GSH and obstructing the expressions of TGF-β and TNF-α [129].
Additionally, some flavonoids and polyphenols have been found to perform or moderate the AIM2 inflammasome pathway. Quercetin treatment was found to impede NLRP3 inflammasome and the expression of pro-caspase-1 as well as suppressing the expression of AIM2 in a dose-dependent fashion in in vitro and in vivo investigations [130, 131]. Treatment with EGCG failed to decrease AIM2 expression, but it could down-regulate the expression of active caspase-1 and IL-1β in epidermal keratinocytes [132]. Several flavonoids have been demonstrated to have multiple inhibiting effects on different inflammasomes. Glycyrrhizin, one of the triterpenoids, has hindering effects on both NLRP3 and AIM2 inflammasomes to decrease the formation of active-form caspase-1 and IL-1β release [133]. Isorhamnetin can impede AIM2 and NLRP3 inflammasome to decrease several pro-inflammatory cytokine expressions, such as IL-1α, β, IL-18, TGF-β and hyperoside; it suppresses the activation of AIM2 and NLRC4 inflammasome but does not affect cytokine expression [134]. In vitro, THP-1 cells treated with apigenin significantly inhibit NLRP3 and AIM2 inflammation to reduce caspase-1 and IL-1β production [135]. The activation of AIM2 inflammasome expression in A549 and H460 cells could be suppressed by luteolin in a concentration-dependent manner [136]. Some flavonoids can serve as activators of inflammasome, i.e. icariside I and bavachin. Icariside I can promote and regulate bone remodelling and is considered to be a candidate compound for osteoporosis treatment [137]. Bavachin can increase the mRNA levels of oestrogen-responsive genes and serve as the activator of oestrogen receptor [138]. A recent study showed that bavachin and icariside I could selectively exacerbate NLRP3 inflammasome but not NLRC4 or AIM2 to promote the expression and activity of caspase-1, TNF-α and IL-1β and that may cause idiosyncratic hepatocyte toxicity [138, 139].
Alternative treatments via inflammasome-mediation for COPD
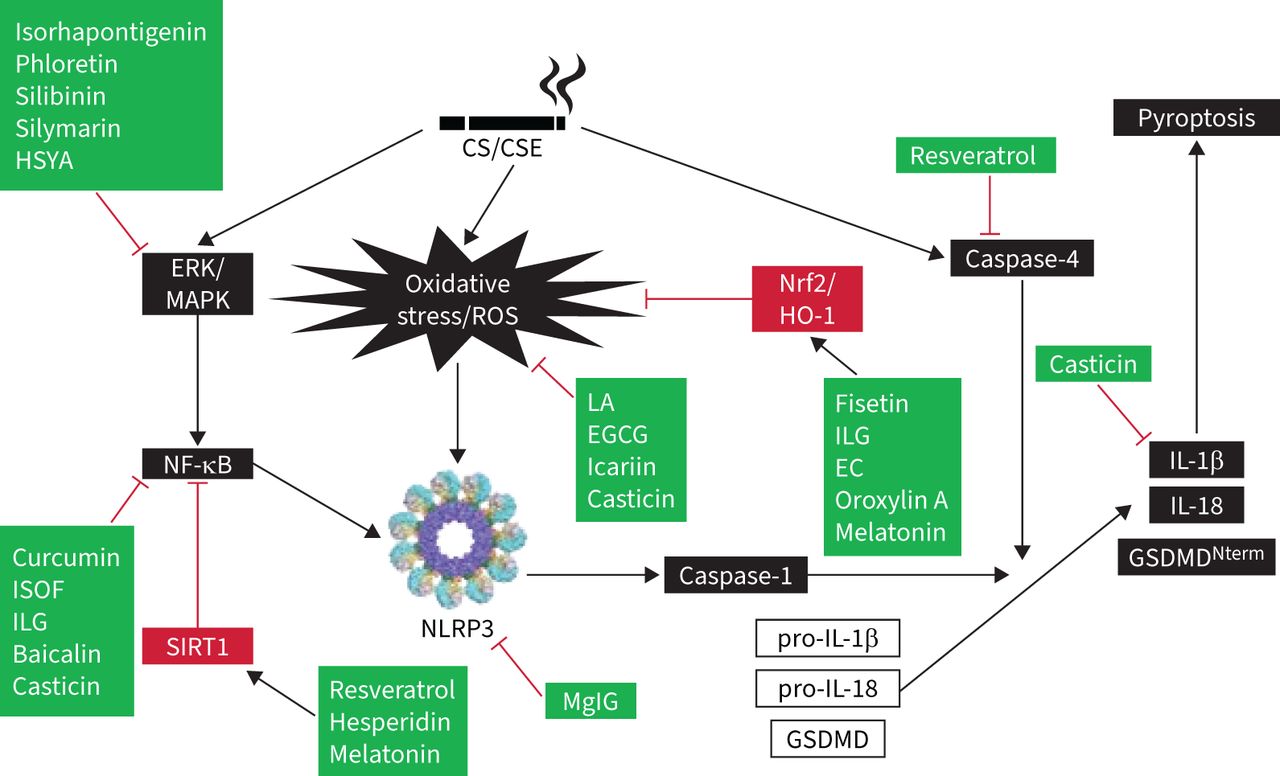
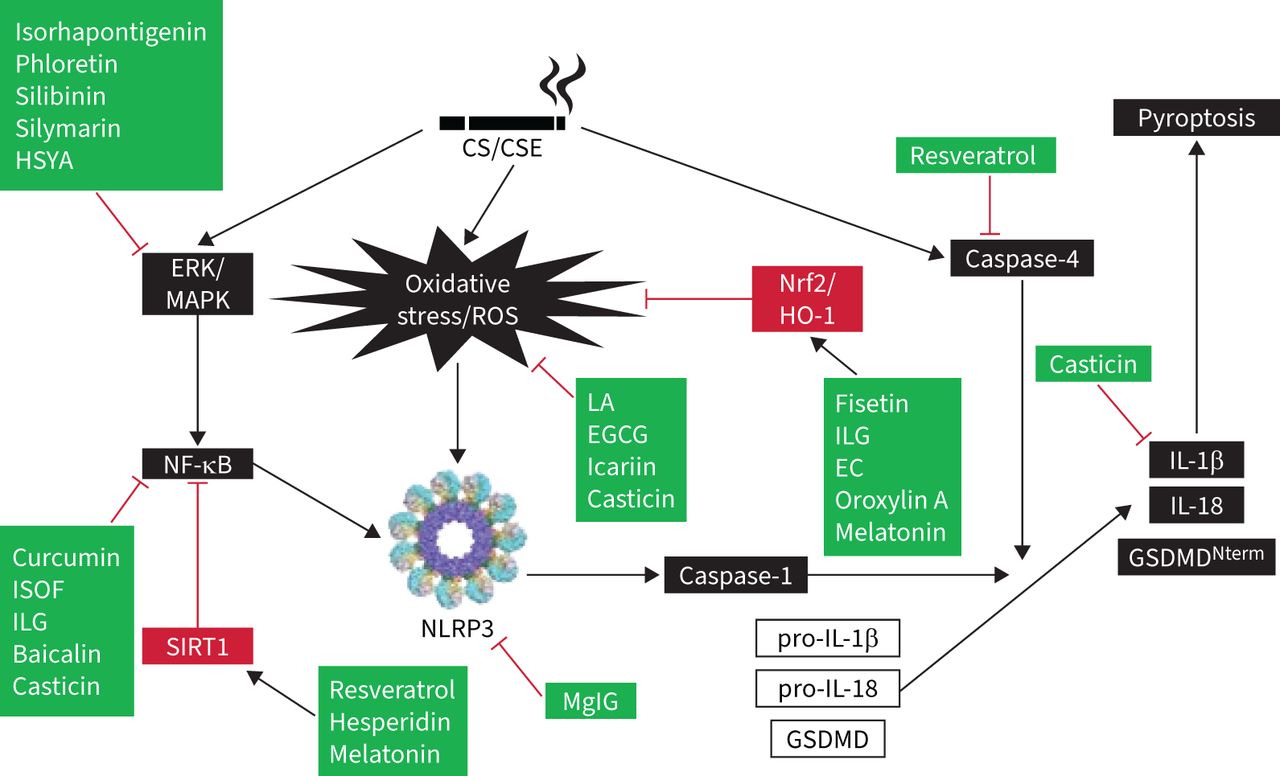
Hydrogen sulphide played a protective role in PM-induced mice emphysema and airway inflammation by hampering the formation of NLRP3 inflammasome and apoptosis via the Nrf2-dependent pathway [140]. Recently, low-molecular-weight anti-inflammatory agents, including antioxidants, inflammasome inhibitors, kinase inhibitors, modulators of inflammatory mediators, phosphodiesterase-4 inhibitors, protease inhibitors, and other agents have shed light on the development of COPD treatment. The molecular docking results of low-molecular-weight agents and targeted proteins provide new insights for targeted COPD treatments, particularly for small-molecule agents [141]. Melatonin showed protection against COPD in vitro and in vivo tests. It activated the intracellular antioxidant thioredoxin-1 (thereby inhibiting the thioredoxin-interacting protein–NLRP3 pathway) and suppressing the impaired mitophagy-mediated inflammasome activation (upregulating PTEN-induced kinase 1, Parkin and LC3B-II expression). Melatonin also enhanced the overall anti-oxidative status of a COPD lung via Nrf2–haem oxygenase-1 axis restoration [142]. Melatonin attenuated airway inflammation via SIRT1-dependent inhibition of NLRP3 inflammasome and IL-1β in rats challenged with CS and LPS [143]. MCC950 is a potent and selective inhibitor of NLRP3 inflammasome and reduced the mRNA expressions of IL-1β, IL-8, TGF-β and MMP-9. At 24 h after LPS instillation, MCC950 could also reduce the protein levels of IL-1β, IL-18 and caspase-1 in mice [144]. Lipoxin receptor agonist (BML-111) may prevent the activation of NLRP3 inflammasome and hinder ROS production via upregulation of Nrf2 in COPD model mice [145]. Forskolin and isoforskolin (ISOF) are polyphenols isolated from Coleus forskohlii and have been reported to have anti-inflammatory and anti-oxidative effects [146]. Forskolin inhibited the activation of NLRP3 inflammasome and decreased the secretions of IL-1β and IL-18 from macrophages [147, 148]. ISOF alleviated the acute exacerbation of COPD by prompting pulmonary function and mitigating inflammation via downregulation of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6), monocyte chemoattractant protein-1, monokine induced by interferon-γ, IP-10, C-reactive protein, Th17/IL-17A and NF-κB/NLRP3 pathways in CS-induced mice [149]. ISOF also tempered IL-1β and IL-18 secretion by NLRP3 inflammasome activation in human macrophages [150]. Treatment with ISOF can downregulate the mTOR level in lung tissues and increase intracellular cyclic adenosine monophosphate levels to relax the histamine-induced contraction of the lungs and trachea smooth muscles. Pre-treatment with ISOF significantly ameliorated the pathological damage to lung tissue and improved pulmonary function in COPD and airway hyper-responsive rats [149, 151]. The differential impact of macrolides-azithromycin on the inflammasome–IL-1β axis may be of consequence in inflammasome-driven diseases, e.g. COPD and asthma [152]. Histidine repressed the activation of NLRP3 inflammasome in both PPE- and LPS-induced COPD mouse models and in vitro mouse alveolar macrophage MH-S cells [153]. Coixol inhibited MAPKs, NF-κB pathways and the activation of NLRP3 inflammasome in LPS-induced RAW 264.7 cells [154]. After long-term PM-exposure-induced lung inflammation and fibrosis, resveratrol intervention improved these adverse effects by preventing autophagy-related NLRP3 inflammasome activation [155]. CS-challenged mice were treated with flavonoids isolated from loquat (Eriobotrya japonica) leaves, the results revealed that flavonoids have a protective effect and putative mechanism of the action of total flavonoids resulted in the inhibition of inflammation and oxidative stress through the regulation of transient receptor potential vanilloid 1 and related signal pathways in lung tissues [156]. Table 2 lists the mediating roles of polyphenols and flavonoids on the amelioration of CS-induced NLRP3 inflammasome in COPD. Additionally, figure 2 illustrates the acting target molecules of the polyphenols and flavonoids on the signal pathways of CS-induced NLRP3 inflammasome. From this diagram, it is obvious that this has been separated into three approaches: the first, CS/CSE, causes the involvement of CS-induced inflammasome signal pathways (shown in black boxes) and the second is the attenuating mediators of regulating signals (shown in red boxes). Thirdly, the acting targets of polyphenols and flavonoids are displayed in green boxes (figure 2). Moreover, quercetogetin suppressed mitophagy-dependent apoptosis by inhibiting the expression of cleaved caspase-3, -8 and -9, and downregulating caspase activity in human lung bronchial epithelial cells (BEAS-2B cells) exposed to CSE [157].
The mediating roles of polyphenols and flavonoids on the amelioration of cigarette smoke (CS)-induced inflammasome in COPD
{kind=link}
{kind=link}
The acting target molecules of polyphenols and flavonoids on the signal pathways of cigarette smoke (CS)-induced inflammasome. Black boxes represent effects of CS/cigarette smoke extract (CSE), caused by the involvement of signal pathways. Red boxes contain the attenuating mediators of regulating signals. Green boxes display the acting targets of polyphenols and flavonoids. Black arrows depict stimulation whereas red “T” bars represent suppression. EC: endothelial cell; EGCG: epigallocatechin-3-gallate; ERK: extracellular signal-regulated kinases; GSDMD: gasdermin D; HO-1: haem oxygenase-1; HSYA: hydroxysafflor yellow A; IL: interleukin; ILG: isoliquiritigenin; ISOF: isoforskolin; LA: liquiritin apioside; MAPK: mitogen-activated protein kinase; MgIG: magnesium isoglycyrrhizinate; NF-κB: nuclear factor-κB; NLRP3: nucleotide-binding oligomerisation domain-like receptor family, pyrin domain containing protein-3; Nrf2: nuclear factor erythroid 2-related factor 2; ROS: reactive oxygen species; SIRT1: sirtuin 1.
Future perspectives
Inflammation plays a vital role in the development of COPD. Pyroptosis, an inflammatory form of cell death, may be implicated in the pathogenesis of COPD. The NLRP3 inflammasome enhances inflammatory cell recruitment and mediates immune responses in the lungs. Further, this is involved in numerous diseases characterised by induced lung disease by focusing on pathways causing chronic respiratory epithelial cell injury, cell death, alveolar destruction and tissue remodelling affiliated with the progress of ALI and COPD. The experimental findings may afford mechanistic insights into the immunosuppression in smokers via the CS-activated ROS–NLRP3 axis and induced epithelial cells pyroptosis. According to the abovementioned section, polyphenols and flavonoids may have therapeutic potential in CS-COPD and present exclusive opportunities to develop an approach or strategy to modulate immune functionality via mediating NLRP3 inflammasome. Corticosteroids are the most-used anti-inflammatory agent for asthma and COPD. However, people with severe asthma or COPD show poor response to the anti-inflammatory effects of corticosteroids. The Corticosteroid resistance is a main therapeutic challenge to the management of COPD. Prominently, resveratrol, icariin and quercetin could restore the sensitivity of corticosteroid, and, in combination with corticosteroids, have the potential to form novel treatments for COPD. A panel of clinically effective drugs has displayed potential in restoring steroid- resistance in experimental models. It is highly plausible that some of these phytocompounds or molecules can be efficaciously repositioned for clinical use in the management of COPD. One clinical trial has reported that 21 COPD patients (forced expiratory volume in 1 s (FEV1) 53±15% predicted; age 67±9 years; body mass index 24.5±3.3 kg·m−2) received resveratrol (150 mg·day−1) or placebo for 4 weeks, the results failed to ratify the previously designated positive effects of resveratrol on the mitochondrial function of skeletal muscle in patients with COPD [158]. In mild-to-severe lung disease, COPD patients with FEV1 ranging between >35% and <80% were recruited and supplemented with either quercetin (500∼2000 mg·day−1) or placebo for 1 week. The patients showed no drug-related severe adverse manifestations based on blood tests, which contained complete blood counts and an evaluation of a comprehensive metabolic panel [159].
Questions for future research
Remarkably, underlying mechanisms and some possible causes have led to inconsistent results in observational studies and supplementation trials because investigations in this field are mostly limited to animal models or small clinical trials. More forthcoming cohorts and well-designed clinical trials are needed to support the introduction of individualised phytochemical interventions into health policy.
Points for clinical practice
Recently, NLRP3 inflammasome has been confirmed to play a significant role in the pathogenesis of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection and is involved in the cytokine storm formation of ALI associated with coronavirus disease 2019 (COVID-19). Up to date, there are more than 1300 scientific articles that have looked into the link between COPD and COVID-19, and more than 280 reports have explored the inflammasome mechanisms in the pathogenesis of COVID-19. Notably, there is accumulating evidence that the inducers, components, pathways and effectors of inflammasome signal pathways are attributed to the pathogenesis in COPD and SARS-CoV-2 infection [160, 161]. On the other hand, SARS-CoV-2 infection may cause more severity in COPD patients [162]. A clinical report found that curcumin can mediate and down-regulate the NLRP3 inflammasome relative intracellular signal transduction pathways that are involved in inflammation of COVID-19 patients [163]. This is an electrifying outcome for combating COVID-19 and creates a new hope for promoting drug development from the medicinal perspective of curcumin.
Footnotes
Provenance: Submitted article, peer reviewed.
Author contributions: N. Kang, Y. Yu and Y. Mi participated in paper collection and data analysis. Y-S. Fu, J. Guo and J. Wu did manuscript draft preparation. Y-S. Fu and C-F. Weng critically discussed and reviewed the manuscript. All authors have reviewed and approved the final version of manuscript for submission and publication.
Conflict of interest: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest with regard to the content of this report.
Support statement: This work is financially supported by Xiamen Medical College for C-F. Weng (K2019-01).
- Received February 12, 2022.
- Accepted April 20, 2022.
- Copyright ©The authors 2022
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References











Jump To
- Article
- Abstract
- Abstract
- Introduction
- Inflammasome
- Polyphenols and flavonoids
- The pathophysiology of CS-COPD
- Polyphenols treatment via inflammasome in a cigarette smoking model
- Alternative treatments via inflammasome-mediation for COPD
- Future perspectives
- Questions for future research
- Points for clinical practice
- Footnotes
- References
- Figures & Data
- Info & Metrics