





Abstract
Neutrophilic inflammation has a key role in the pathophysiology of multiple chronic lung diseases. The formation of neutrophil extracellular traps (NETs) has emerged as a key mechanism of disease in neutrophilic lung diseases including asthma, COPD, cystic fibrosis and, most recently, bronchiectasis. NETs are large, web-like structures composed of DNA and anti-microbial proteins that are able to bind pathogens, prevent microbial dissemination and degrade bacterial virulence factors. The release of excess concentrations of proteases, antimicrobial proteins, DNA and histones, however, also leads to tissue damage, impaired mucociliary clearance, impaired bacterial killing and increased inflammation. A number of studies have linked airway NET formation with greater disease severity, increased exacerbations and overall worse disease outcomes across the spectrum of airway diseases. Treating neutrophilic inflammation has been challenging in chronic lung disease because of the delicate balance between reducing inflammation and increasing the risk of infections through immunosuppression. Novel approaches to suppressing NET formation or the associated inflammation are in development and represent an important therapeutic target. This review will discuss the relationship between NETs and the pathophysiology of cystic fibrosis, asthma, COPD and bronchiectasis, and explore the current and future development of NET-targeting therapies.
Abstract
NETs contribute to the pathophysiology of chronic lung disease. Immunomodulating therapies that may reduce inflammatory mediators and NET formation, without compromising bacterial clearance, offer a new treatment path for patients. https://bit.ly/3fyJC6I
Introduction: neutrophil extracellular traps (NETs) – structure and function
Chronic lung conditions such as chronic obstructive pulmonary disease (COPD), asthma and bronchiectasis represent a large healthcare burden, affecting an estimated 545 million people worldwide [1].
While each condition has a distinct pathophysiology, they share important characteristics, including the presence of airway inflammation, airflow obstruction, airway remodelling and mucociliary dysfunction. There is substantial overlap in the clinical characteristics of these conditions and many patients have features of more than one pathology. Historically, airway inflammation in asthma was considered to be primarily eosinophil driven; with COPD, bronchiectasis and cystic fibrosis (CF) being thought of as primarily neutrophil driven. Considerable heterogeneity in airway inflammatory phenotypes is now recognised with the subtypes of neutrophilic asthma, and eosinophilic COPD and eosinophilic bronchiectasis now being identified.
Treatments for airways disease typically consist of inhaled corticosteroids and bronchodilators to treat eosinophilic airway disease and bronchoconstriction, respectively. Oral, intravenous or inhaled antibiotics are used to treat airway bacterial infection, and mucoactive drugs and physiotherapy may be used to manage symptoms of mucus obstruction.
Neutrophilic inflammation, however, remains a feature of chronic airways diseases that has proven most challenging to understand and to therapeutically target. Identifying the mechanisms leading to neutrophilic inflammation and finding therapies targeting this process is critical. Up to 50% of symptomatic asthma patients [2–4], 60–80% of COPD patients [5–7] and 70–90% of bronchiectasis patients [3, 8, 9] may be classified as neutrophilic and therefore currently lack a direct anti-inflammatory treatment approach.
This review discusses recent advances in our understanding of neutrophilic inflammation in chronic airways diseases, with a focus on the emerging role of neutrophil extracellular traps (NETs). We will discuss the associations between NETs and disease burden in different conditions, and the potential for new and repurposed therapies to target NETosis including novel anti-inflammatory and immunomodulatory treatments.
Neutrophils
Neutrophils are the among the first immune cells recruited to the lungs following infection. These cells are generated in the bone marrow with subsequent deployment to the bloodstream for transport to tissues along a chemotactic gradient [10]. Neutrophils utilise a number of host defence mechanisms, including phagocytosis, degranulation, production of reactive oxygen species (ROS) and reactive nitrogen species, pro-inflammatory cytokine production, and NET formation. These complex and diverse mechanisms give neutrophils the flexibility to respond to a range of foreign antigens, including bacteria, viruses and fungi.
First identified in 1996 by Takei et al. [11], NETs were originally thought to be another programmed method of cell death following stimulation with phorbol 12-myristate 13-acetate (PMA), with a different molecular pathway from apoptosis and necrosis. In 2004, NETs were identified as a neutrophil response able to kill bacteria in vitro, and the process was named “NETosis”. Neutrophils stimulated with either PMA or C-X-C motif ligand 8 (CXCL-8) extruded extracellular fibres composed of nuclear and granule constituents. These extracellular structures were able to bind Gram-positive and Gram-negative bacteria, prevent micro-organisms spreading, and were able to degrade bacterial virulence factors [12]. Despite these findings, whether NETs are truly antimicrobial, or trap pathogens and prevent infection from being disseminated in vivo, remains controversial.
Lytic NETosis, a cell death pathway, is initiated through ligand binding to neutrophil complement and toll-like receptors [12–15]. Upon activation, receptors on the neutrophil surface initiate an RAF/mitogen-activated protein (MAP)/extracellular signal-regulated kinase (ERK) kinase (MEK)/ERK (RAF/MEK/ERK) cascade resulting in the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and superoxide production [16]. Hydrogen peroxide causes azurosome granules to release neutrophil elastase (NE) and other serine proteases, as well as initiating the production of ROS [17]. Under the influence of ROS, granules and the nuclear envelope rupture, and peptidyl arginine deiminase 4 (PAD4) is activated [18]. Released NE and myeloperoxidase (MPO) migrate to the nucleus and cleave histones as activated PAD4 citrullinates histones, leading to chromatin decondensation [19]. PAD4 [20], NE [17] and MPO [21] are all critical to NET formation, which can be reduced through the inhibition of any of these components. In the final step, the plasma membrane ruptures, and the web-like NET is released, resulting in cell death (figure 1) [22].

Neutrophil extracellular trap (NET) formation. a) NET formation is activated through a variety of stimuli. Upon activation, receptors on the neutrophil surface initiate a RAF/mitogen-activated protein (MAP)/extracellular signal-regulated kinase (ERK) kinase (MEK)/ERK (RAF/MEK/ERK) cascade resulting in the production of nicotinamide adenine dinucleotide phosphate oxidase and superoxide production. b) Hydrogen peroxide (H2O2) causes the release of neutrophil elastase (NE) and other serine proteases from the azurosome. These proteases move into the nucleus and cleave histones. H2O2 production of reactive oxygen species activates peptidyl arginine deiminase 4 (PAD4), leading to the citrullination of histones. c) The nuclear envelope breaks down and granular proteins mix with DNA. The plasma membrane ruptures, and the NET is released into the extracellular space.
In 2007, Clark et al. [23] described a rapid, novel mechanism of NET formation, termed “vital NETosis”. During this process, neutrophils remain viable despite the formation of NET structures. Briefly, condensation of the multi-lobed nucleus is followed by separation of the inner and outer nuclear membranes and budding vesicles. Nuclear DNA fills the separated membrane vesicles, which are then extruded into the extracellular space before rupturing and releasing chromatin. During vital NETosis, the integrity of the plasma membrane remains intact [23, 24], unlike lytic NETosis where rupturing of the plasma membrane leads to cell death. There is also evidence to suggest that neutrophils undergoing vital NETosis are also able to retain chemotaxis and phagocytic activity [25].
NETs have a characteristic web-like structure and there are several methodologies used to quantify NETs both in vitro and in vivo, including immunolabelling, electron microscopy and immunoassays for NET complexes such as DNA–MPO, DNA–elastase and histone–elastase. NETs are a highly ordered, branching filament network of DNA and antimicrobial proteins, similar to the “beads on a string” model of nucleosome chromatin [26]. Cross-linking of NET proteins through chlorinated polyamines and polyamide bonds ensures that NET formation is both ordered and reproducible [27]. The major structural components of NETs make up part of their defence against invading pathogens. The phosphodiester backbone of NETs contributes to antimicrobial activity through cation chelation, lysing of bacterial cells and disruption of membrane integrity [28]. NETs are complex structures and proteomic analysis has identified 330 NET component proteins [29]. A host of antimicrobial granular and cytoplasmic proteins have been identified as part of the web-like structure, including NE, MPO, cathepsin G, proteinase 3, LL-37 and calprotectin [17, 30–33]. NET formation is heterogenous and can be induced by a range of biological and synthetic stimuli, including PMA, lipopolysaccharides (LPS), bacteria, cigarette smoke and some environmental factors. Petretto et al. [29] utilised proteomic analysis to study the protein composition of NETs produced after stimulation with PMA, calcium ionophore A23187, Escherichia coli LPS or untreated. They found that there was a common core of component NET proteins; however, under each stimulus differentially expressed proteins were also detected. PMA- and A23187-induced NETs were similar, whereas LPS-induced and spontaneous NETs widely diverged in protein composition. Additionally, post-translational modifications varied under different stimuli. Methionine sulfoxidation was the most frequent modification, especially in PMA- and LPS-induced NETs and MPO was the more extensively modified protein. As different stimuli can affect the protein composition and post-translational modifications involved in NET formation, it is likely that NETs induced under different stimuli may have different biological functions [29, 34–36].
Persistence of NETs at inflammatory sites due to impaired NET degradation is also thought to be associated with autoimmunity [37]. Anti-neutrophil cytoplasmic antibodies (ANCAs) target neutrophil proteins such as MPO and proteinase 3. Studies have demonstrated that ANCAs activate neutrophils by inducing respiratory burst and triggering NET formation [38]. NETs have been implicated in the pathogenesis of ANCA-associated vasculitis, a disease in which systemic necrotising vasculitis predominantly affects the small vessels in the lungs and kidneys [39].
NETs have been described as part of the pathophysiology of a number of chronic lung diseases, where they have been implicated in local and systemic tissue damage, mucus plugging, and disruption of the immune response, propagating the cycle of inflammation. The following sections will discuss these implications in more detail in relation to CF, COPD, asthma and bronchiectasis.
NETs and inflammation in chronic lung disease
CF
NETs have been extensively studied in CF. CF sputum has been shown to be heavily composed of NET and NET-related proteins, including NE, MPO, calprotectin and resistin [40–45]. NE, which has long been recognised as a biomarker of disease severity in CF and is believed to contribute to disease progression, retains its catalytic ability when bound to DNA and has been shown to have elevated protease activity within the extracellular DNA (eDNA) of sputum from CF patients [42]. Additional NET-associated markers such as resistin and MPO are associated with worse disease outcomes and reduction in lung function in CF [41, 46]. Calprotectin (S100A8/A9) is an NET protein that has been found in sputum [47], serum [48] and bronchoalveolar lavage (BAL) fluid [49] of patients with CF. Serum calprotectin levels can be used as a biomarker in CF patients to predict time to next exacerbation and lung function decline [45]. Autoantibodies directed against bactericidal permeability-increasing protein (BPI) have been implicated in the pathogenesis of CF and are associated with a reduction in lung function [50]. BPI mediates extracellular bactericidal and LPS-neutralising functions through high-affinity binding of LPS on the bacterial envelope. A study by Skopelja-Gardner et al. [51] proposed that the breaking of tolerance to BPI is facilitated by Pseudomonas aeruginosa and that P. aeruginosa-mediated NET formation results in BPI cleavage by P. aeruginosa elastase. This study provides an insight into the development of autoimmunity to BPI and the role that NET formation plays.
The excessive NET formation found in CF sputum contributes to its viscosity and gel-like structure [40]. Higher levels of free eDNA are found in CF sputum compared to BAL fluid and lung tissue, suggesting that neutrophilic inflammation is more dominant in the proximal airways. Previous studies in CF have shown that increased airway eDNA levels are associated with neutrophilic inflammation, lung function decline, increased chemokine levels and fungal colonisation [46, 52, 53]. A 2014 study identified macrophage migration-inhibitory factor (MIF) as a regulator of NET formation in CF and that MIF levels correlated with a reduction in lung function [54]. Additionally, NETs have been shown to prime macrophages for cytokine release, suggesting that the interplay between macrophages and NETs contributes to the chronic inflammation associated with CF [44, 55]. These studies indicate that NETs significantly add to the burden of disease, but do not fully explain why NETs occur in high levels in the most severely affected patients. Typically, neutrophils undergo spontaneous apoptosis as part of the response to infection and inflammation [56]. Delayed apoptosis has been previously described in CF and is thought to contribute to neutrophilic inflammation in the lung [57]. A study by Gray et al. [44] found that due to decreased apoptosis, CF neutrophils had a longer life span, which allowed for increased NET production. This could be reversed by the CF transmembrane conductance regulator potentiator ivacaftor in patients with G551D mutations. It is evident that NETs add to the burden of disease in CF through several mechanisms, including structural changes to sputum, increasing levels of inflammatory proteins, and interaction with other inflammatory cells leading to a self-perpetuating cycle of inflammation. In a recent study, MPO inhibition in mice was found to improve morbidity by limiting oxidative stress, without compromising bacterial clearance [43]. NE-knockout mice have been shown to have significantly reduced airway neutrophilia, mucus hypersecretion, goblet cell metaplasia and structural lung damage [58]. These studies suggest a key role for NETs and component proteins in CF lung disease and that therapeutically targeting NETs may have potential clinical benefits.
COPD
Smoking results in an increase in neutrophilic inflammation, a hallmark of inflammation in COPD [59]. COPD patients have raised inflammatory markers and prominent airway neutrophilic inflammation compared to healthy controls, with an association between sputum neutrophilic inflammation and Global Initiative for Chronic Obstructive Lung Disease severity classification [7]. Additionally, raised blood neutrophil counts have been correlated with exacerbation frequency and mortality in patients with COPD [6]. Alongside chronic neutrophilic inflammation, NETs have also been found in the airways of both stable and exacerbating patients with COPD [60] and in murine models have been shown to be induced by cigarette smoke [35]. A study of 99 subjects with COPD found that sputum NETs, measured by immunoassay, were associated with exacerbation frequency and reduction in forced expiratory volume in 1 s (FEV1) [61]. A number of NET proteins have been identified in the airways of COPD patients and are believed in contribute to inflammation and tissue damage, including NE, MPO and leukotriene B4 (LTB4) [62, 63]. Inflammatory markers in clinically stable moderate COPD have good repeatability over time, making them robust targets for inflammatory marker monitoring [64]. NE, a key component of NETs, is associated with disease severity and bacterial infection in COPD [63]. A 2013 study by Jiang et al. [62] demonstrated that NE impairs epithelial defence against non-typeable Haemophilus influenzae infection through the degradation of airway epithelial host defence proteins, making patients more susceptible to bacterial infection. Levels of NET components, such as eDNA, LL-37 and NE, are elevated in COPD patients compared to healthy controls along with the pro-inflammatory cytokines CXCL-8 and interleukin (IL)-1β [65]. Furthermore, PAD4 gene expression is significantly higher in neutrophilic compared to non-neutrophilic COPD [65].
Previous studies have suggested that CXCL-8 induces NETosis via C-X-C motif chemokine receptor (CXCR) 1 and CXCR2 on the surface of neutrophils. A study from 2018 showed that sputum from COPD patients was capable of inducing NETosis in peripheral blood neutrophils of healthy donors and treatment with a selective CXCR2 antagonist significantly reduced NET formation [15, 66]. However, our recent study did not find an effect of the reversible CXCR2 antagonist danirixin on NET formation in vivo in a pilot randomised trial [67].
During exacerbations of COPD, the chemoattractant LTB4 contributes to the neutrophil influx in the airway and may influence disease progression [68]. Moreover, markers of neutrophilic inflammation increase during acute exacerbations of COPD independently of a detectable viral or bacterial airway infection [69]. Previous studies have reported that alveolar macrophages exhibit defects in their phagocytic ability, reducing the clearance of apoptotic airway cells [70]. High levels of pro-inflammatory cytokines and neutrophil chemokines can result in a positive feedback loop, contributing to the persistent number of airway neutrophils seen in COPD. Coupled with the reduced clearance of cells by macrophages, this evidence may partially account for the elevated levels of NETs in the airways of COPD patients. The cytotoxic properties of NET components and their reduced antimicrobial function may partly explain their association with disease severity, airflow limitation and microbial dysbiosis [60, 61, 71].
Asthma
Historically, asthma has been considered an eosinophilic disease; however, more recent studies have identified a subtype of severe patients with neutrophilic inflammation. Neutrophilic asthma has been associated with increased inflammatory markers, bacterial burden and microbiome dysbiosis [32, 72]. Isolated peripheral blood neutrophils of severe asthma patients have enhanced production of IL-17A, IL-17F and signal transducer and activator of transcription 3 phosphorylation, indicating that neutrophil infiltration of the lungs may represent a source of pro-inflammatory cytokines which provide a positive feedback loop that sustains inflammation [73].
Neutrophil and NET proteins such as NE and calprotectin have been detected in the sputum and serum of asthma patients, respectively [74–76]. In mice with established airway disease, inhibition of NE reduced airway hyperresponsiveness (AHR), goblet cell metaplasia and inflammatory cell accumulation, indicating that NE is involved in the development of allergic airway inflammation [75]. Calprotectin levels in serum are increased in patients with asthma, and correlate with blood neutrophil percentage, AHR and lung function [74].
A 2019 study investigated the role of activated neutrophils and inflammasome activation in 399 patients with severe asthma and 94 healthy controls [77]. eDNA was measured in the induced sputum of subjects and asthma patients were stratified into eDNA-low and -high subgroups, representing those with high and low levels of NETosis respectively. NE–DNA complexes and citrullinated histone H3–DNA complexes, which are more specific markers of NET formation, were increased in eDNA-high patients but not in eDNA-low patients. Patients in the eDNA-high subgroup had lower Asthma Control Test scores, frequent use of oral corticosteroids and frequent history of chronic mucus hypersecretion compared to their eDNA-low subgroup counterparts. As is seen in COPD, eDNA levels were raised in the sputum of severe asthma patients and were associated with elevated CXCL-8, IL-1β and caspase-1 activity [65, 77]. This fits with the observation that NETs are able to activate the inflammasome in cells such as monocytes and macrophages, triggering the secretion of IL-1β [77–80]. The inflammasome is an intracellular molecular scaffold that promotes the cleavage and secretion of IL-1β and IL-18 through activated caspase-1. The release of IL-1β promotes neutrophil recruitment to that lung, further perpetuating the cycle of inflammation. Neutrophil recruitment by NETs is also amplified by the NETotic stimulation of airway epithelial cells to express CXCL-1, CXCL-2 and CXCL-8 via the Toll-like receptor 4/NF-κB pathway [81].
Rhinovirus infection is a key trigger of asthma exacerbations and may trigger both neutrophilic and eosinophilic immune responses. A study by Toussaint et al. [82] published in 2017 investigated the relationship between respiratory viral infections and type 2 immune responses. 28 subjects with mild-to-moderate asthma and 11 healthy volunteers were experimentally infected with rhinovirus (RV) 16 and nasal lavages were performed at multiple timepoints across a 2-week period. Nasal double-stranded (ds)DNA was increased in both groups following RV infection, with peak concentrations of dsDNA being higher in the asthma subjects. Furthermore, type 2 cytokines IL-5 and IL-13 were raised in the asthma subjects, who also reported greater upper and lower respiratory symptom score than the healthy control subjects. Nasal concentrations of dsDNA correlated positively with cold symptom severity and asthma exacerbation severity and were associated with lower airway type 2 response. This demonstrates that in subjects with asthma, RV infection induces dsDNA release correlating with type 2 immune-mediated asthma exacerbation severity. The authors used a murine model to further investigate the relationship between RV-mediated release of dsDNA and asthma exacerbation. They found that RV infection triggers a dsDNA release associated with NET formation and that NET release can be reduced through blocking NE or degradation through deoxyribonuclease (DNase) treatment. Finally, mice were administered with endogenous dsDNA, which triggered a type 2 immune response, demonstrating that NET formation contributes directly to T-helper cell type 2-mediated inflammation. These effects on inflammatory cells are in addition to the well-recognised NET-mediated damage to epithelial cells in vitro, suggesting that NETs contribute to the pathophysiology of asthma through multiple mechanisms [83].
Bronchiectasis
One of the major hallmarks of bronchiectasis is non-resolving neutrophilic inflammation. Cole et al. [84] described bronchiectasis as a vicious cycle of bacterial colonisation, neutrophilic inflammation, structural airway changes and impaired mucus clearance. This perpetual cycle results in chronic inflammation and tissue damage, furthering the progression of the disease. A review in 2018 described the interactions of these four steps as a “vicious vortex”, where each element can have a significant impact on another, resulting in a persistent and progressive process over time [85]. The vortex model explains why previous “breaking the cycle” treatments, such as antibiotics, have not had a significant effect on the clinical outcomes for patients. Despite treating one aspect of the vortex, the other components can continue to act on each other, sustaining the disease progression. Treating multiple components of the vortex simultaneously may lead to better outcomes for patients.
Neutrophilic inflammation is a key mediator in both the cycle and vortex models of bronchiectasis. A study of 152 patients with stable bronchiectasis demonstrated that sputum neutrophilic inflammation correlated with severity, duration of disease and a reduction in pulmonary function as measured by FEV1 % [86]. Bacterial colonisation is believed to be the main stimulant of neutrophil migration into the airway in bronchiectasis. It has been demonstrated that there is a direct correlation between bacterial load and markers of airway inflammation, and that treatment with short- and long-term antibiotic therapies reduced markers of airway and systemic inflammation [87]. A separate study by Angrill et al. [88] evaluated differential cell counts, quantitative bacteriologic cultures and inflammatory mediators in BAL samples from 49 stable patients and nine control subjects. Patients with bronchiectasis had significantly higher concentrations of NE, MPO, tumour necrosis factor alpha (TNF-α), CXCL-8 and IL-6 compared to control subjects, and this raised inflammatory profile correlated with a higher bacterial load. However, this study also identified a subset of patients for whom inflammation occurred despite an absence of bacterial infection.
NE is a biomarker of disease severity in bronchiectasis and is thought to contribute to the pathogenesis of the disease through slowing ciliary beat frequency and stimulating mucus secretion [89, 90]. Raised NE activity in sputum is associated with severity, exacerbation frequency, reduction in FEV1 % predicted and radiological extent of bronchiectasis [9, 91]. Patients with more severe bronchiectasis have been shown to have higher sputum levels of the NET-associated pro-inflammatory mediators lactoferrin and cathelicidin LL-37 and lower levels of the anti-inflammatory secretory leucocyte protease inhibitor (SLPI). Additionally, higher LL-37, a known NET protein, and lower SLPI levels were associated with lower FEV1, P. aeruginosa infection and a shorter time to next exacerbation, demonstrating that patients with bronchiectasis have dysregulated antimicrobial peptide levels which are associated with disease severity [92]. This may be partly explained by the inhibitory function of SLPI on NE, which has been shown to reduce NET formation in murine models [93].
As seen in CF, BPI autoimmunity is associated with P. aeruginosa infection in patients with bronchiectasis. This relationship between P. aeruginosa and BPI autoantibodies may hinder the bactericidal and anti-inflammatory functions of BPI, increasing P. aeruginosa colonisation [51]. If NET formation is involved in BPI disruption in bronchiectasis, as has been shown in CF, anti-NET treatments may reduce chronic infections of P. aeruginosa. A recently published study from our group identified a subgroup of severe patients with bronchiectasis characterised by elevated NET proteins. Sputum NET levels were measured in UK and European cohorts totalling 450 patients and were found to be associated with exacerbation, reduced time to next exacerbation, reduced lung function, worsening quality of life and increased mortality. Additionally, higher NET levels were associated with microbial dysbiosis and a dominance of the Proteobacteria genera Pseudomonas and Haemophilus. In two cohorts of 20 patients, antibiotic treatment reduced NET sputum levels and upregulated anti-protease defence; however, this effect was blunted in patients with P. aeruginosa infection. Macrolides, which lack direct antimicrobial activity against P. aeruginosa, were shown to reduce NET levels and improve patient clinical outcomes, suggesting a novel, immunomodulatory effect. Treatment studies that reduce inflammation and improve clinical outcomes demonstrate that NETs are not only a marker of disease severity, but also actively contribute to lung inflammation. These studies demonstrate for the first time that NETs are associated with the burden of disease in bronchiectasis and that treatment response is linked to reducing NET levels through macrolide or antibiotic therapies [94].
NETs and infection
Bacterial infection
A hallmark of many chronic lung diseases is reduced microbial diversity; however, the mechanisms behind why this happens are not fully understood. Longitudinal cohort studies in both COPD and bronchiectasis have found that microbiome profiles dominated by Proteobacteria are correlated with an increase in severity [61, 94]. In COPD, Haemophilus-associated immune responses have been correlated with neutrophilic inflammation, implicating interactions between host immune response and the microbiome [95]. Furthermore, NET formation has been demonstrated to be associated with an increase in pathogenic Proteobacteria in both bronchiectasis and COPD [61, 94]. In a longitudinal cohort study of 99 subjects with COPD, increasing NET levels were associated with a decreasing Shannon–Weiner diversity index (SWDI), an alpha diversity measure of evenness and richness, and a clear relationship between Haemophilus and NET levels was established [61]. In both studies, Proteobacteria dysbiosis was associated with an increase in severity. In a study of 175 patients with bronchiectasis, high NET levels were associated with a loss of alpha-diversity, as measured by the SWDI and the Chao index, and a significant change in beta-diversity. Patients with high NET levels had an increase in Proteobacteria, specifically Pseudomonas and Haemophilus. Antibiotic treatment in a separate cohort reduced levels of sputum NET proteins, suggesting that certain pathogens are implicated in NET formation [94].
LPS, found in the outer membrane of Gram-negative bacteria, has been demonstrated to stimulate NET formation. Neutrophils are able to recognise LPS from different bacterial sources with the presence of platelets determining NETotic outcome. In the presence of platelets, LPS serotypes from P. aeruginosa, Salmonella enterica and various E. coli strains stimulate vital NETosis. However, under serum- and platelet-free conditions, in a mock tissue model, neutrophils selectively release suicidal NETs in response to LPS derived from only P. aeruginosa and specific E. coli strains [96]. An array of pathogens that can be found in the airways have been shown to induce NETosis, including S. aureus [24], H. influenzae [97], P. aeruginosa [98] and Mycobacterium tuberculosis [99].
One of the most well-characterised pathogens stimulating NETosis is P. aeruginosa, which has been extensively studied in CF patients. CF patients chronically infected with P. aeruginosa have a higher abundance of NET-related proteins in sputum compared to healthy control subjects [100] and both laboratory and CF isolate strains of P. aeruginosa have been shown to induce NADPH oxidase-dependent NET formation [101]. Yoo et al. categorised clinical isolates of P. aeruginosa cultured from sputum or oropharyngeal swabs as either “early” or “late” based on the course of each patient's infection. Early isolates induced significantly higher NET levels compared to corresponding late isolates [98]. Furthermore, Martínez-Alemán et al. [102] found that CF isolates stratified by disease severity could induce different NET morphologies. Isolates from severe patients induced spread morphologies, whereas isolates from mild and moderate patients generated globular NET formation morphologies.
A number of methods have been proposed for the induction of NET formation by P. aeruginosa. Pyocyanin, a redox-actived virulence factor secreted by Pseudomonas, promotes NET formation through Jun N-terminal kinase and phosphatidylinositol 3-kinase [103]. Floyd et al. [104] found that early exponential growth phase cultures of P. aeruginosa induced the highest levels of NET release. Additionally, immotile P. aeruginosa lacking a flagellum or with a non-functional flagellum were weak NET inducers, demonstrating that a functional flagellum is essential for Pseudomonas-induced NETosis. This study may explain why early CF isolates are more potent NET inducers; as over the course of CF lung disease P. aeruginosa downregulates flagellum expression [105].
The ability of P. aeruginosa strains to induce NET formation is likely to also provide a survival advantage to the pathogen. A number of studies have shown that P. aeruginosa is resistant to NET killing [28, 106]. Late-CF isolates of P. aeruginosa have been demonstrated to be resistant to NET-mediated killing, correlating with the development of a mucoid phenotype, suggesting that colonisation of the CF airway by P. aeruginosa is an adaptive process [106]. P. aeruginosa secretes extracellular DNases in order to evade the antimicrobial activity of NETs. NETs induce a DNase-phosphatase operon, triggering the secretion of DNase and a predicted alkaline phosphatase, which contribute to the degradation of NETs and reduced NET-mediated killing [107]. The production of DNase disrupts the structural integrity of NETs and neutralises that antimicrobial activity of DNA, while also diluting the concentrations of cytotoxic NET-bound proteins [107, 108]. A number of other pathogens, including S. aureus, group A Streptococcus and Streptococcus suis have all been shown to deploy nucleases as an NET evasions strategy [109–111]. These studies show that the microbiome plays a pivotal role in the inflammatory response and that certain pathogens can drive NET formation through multiple mechanisms. It is possible that the dominance of NET-tolerant organisms in the microbiome of severe patients is due to the selective advantage they have in an NET-rich environment and that inducing further NET formation may benefit their survival.
Viral infection
Viral infection can enhance airway inflammation and lead to NET formation, particularly during exacerbations of chronic lung disease. The discussion of all viral infections is beyond the scope of this review; therefore, the next section will focus on viruses in relation to airway disease exacerbations. Respiratory viral infections are the main cause of exacerbation in asthma [112] and a driver of exacerbation in COPD [113]. RV16 inoculation in adults with allergic asthma increased circulating neutrophils and nasal concentrations of CXCL-8 and granulocyte colony-stimulating factor [112]. Tang et al. [114] recruited 19 subjects with asthma and 28 non-asthmatic controls in order to investigate the effect of respiratory viruses on neutrophils. Isolated peripheral blood neutrophils were stimulated with R848, RV16, respiratory syncytial virus (RSV) or influenza virus. Unlike in the previously described study [82], RV16 and influenza had no effect on cytokine release. However, RSV triggered the release of CXCL-8 and NE from neutrophils and R848 stimulated the release of CXCL-8, NE and matrix metalloproteinase (MMP)-9, where CXCL-8 released was enhanced in neutrophils from asthmatic subjects compared to non-asthmatic controls. As discussed above, RV infections have been demonstrated to induce dsDNA release and NET formation in human and mouse models, respectively [82]. RV-mediated dsDNA release corresponded with an increase in type 2 immune-mediated response and exacerbation severity. NETs have also been detected in the plasma of patients with H7N9 and H1N1 influenza viruses, with levels correlating with severity of disease and systemic inflammatory injury [115]. Interestingly, NETs generated by influenza A are ineffective against secondary bacterial infection, suggesting that virus-induced NETosis may differ functionally from bacteria-induced NETs [116]. NETs have also been identified in the lungs of severely unwell patients with coronavirus disease 2019 (COVID-19). In these patients, high NET concentrations have been associated with the development of acute respiratory distress syndrome, tissue and organ injury, disease severity, and immunothrombosis [117–120]. A study by Veras et al. [121] found that viable severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) induced NET formation in healthy donor neutrophils. Furthermore, SARS-CoV-2-activated neutrophils increased the apoptosis of epithelial lung cells compared to nonactivated neutrophils, demonstrating a role of NETs in the pathophysiology of COVID-19. These studies suggest that viruses, like many other pathogens, are able to induce NET formation which contributes to systemic inflammation and tissue damage.
Fungal infection
There is growing evidence that the lung mycobiome plays a key role in the clinical outcomes of patients with chronic respiratory disease [122, 123]. However, compared to the extensively researched bacteriome, far less is known about the lung mycobiome. A range of fungal pathogens have been identified in the airways of patients with chronic lung disease, with Aspergillus fumigatus and Candida albicans among the most common fungi detected [124], although fungal profiles appear to differ according to lung disease. Patients with CF have been shown to have a dominance of Candida and Malassezia species [125], whereas A. fumigatus, C. albicans and Mycosphaerella tassiana have been identified in patients with asthma [126].
Although there are limited studies on the relationship between the mycobiome and inflammation, A. fumigatus has been shown to induce NET formation. A study by Bruns et al. [127] demonstrated that both human and murine neutrophils produce NETs in response to A. fumigatus. Additionally, the authors imaged NET structures in the lung tissue of mice models infected with A. fumigatus in vivo. Finally, they showed that A. fumigatus conidia escape NET-killing through the surface protein hydrophobin RodA. This study demonstrates that A. fumigatus can trigger NET formation, but is also able to evade NET killing. NETs have calprotectin-dependent antifungal activity against C. albicans, which is lost in calprotectin-deficient animal models [31]. Furthermore, Branzk et al. [128] have demonstrated that neutrophils sense microbe size and release NETs in response to hyphae and other pathogens that are too large to be phagocytosed. Phagocytosis via dectin-1 acts as a sensor of microbe size and downregulates the translocation of NE to the nucleus to prevent NET release. It is likely that, as with bacterial infections, NET formation is a less effective form of fungal killing than phagocytosis. Histone citrullination by PAD4 is a key component of NET formation. Alflen et al. [129] demonstrated that PAD4-deficient mice infected with A. fumigatus had lower fungal burden and less acute lung injury compared to wildtype controls, suggesting that NET inhibition may improve neutrophil function and reduce lung damage. The above studies show that the mycobiome plays a key role in the clinical outcomes of patients and that this may be linked to fungal-mediated inflammation. Further studies are warranted to increase our understanding of the relationship between fungi and the pathogenesis of chronic lung disease. The apparent importance of NET formation in fungal control is an important issue when considering therapies targeting NETosis in chronic lung disease. A simple summary figure illustrating how NETs are linked to inflammation, infection and structural lung damage is shown in figure 2.

{kind=link}
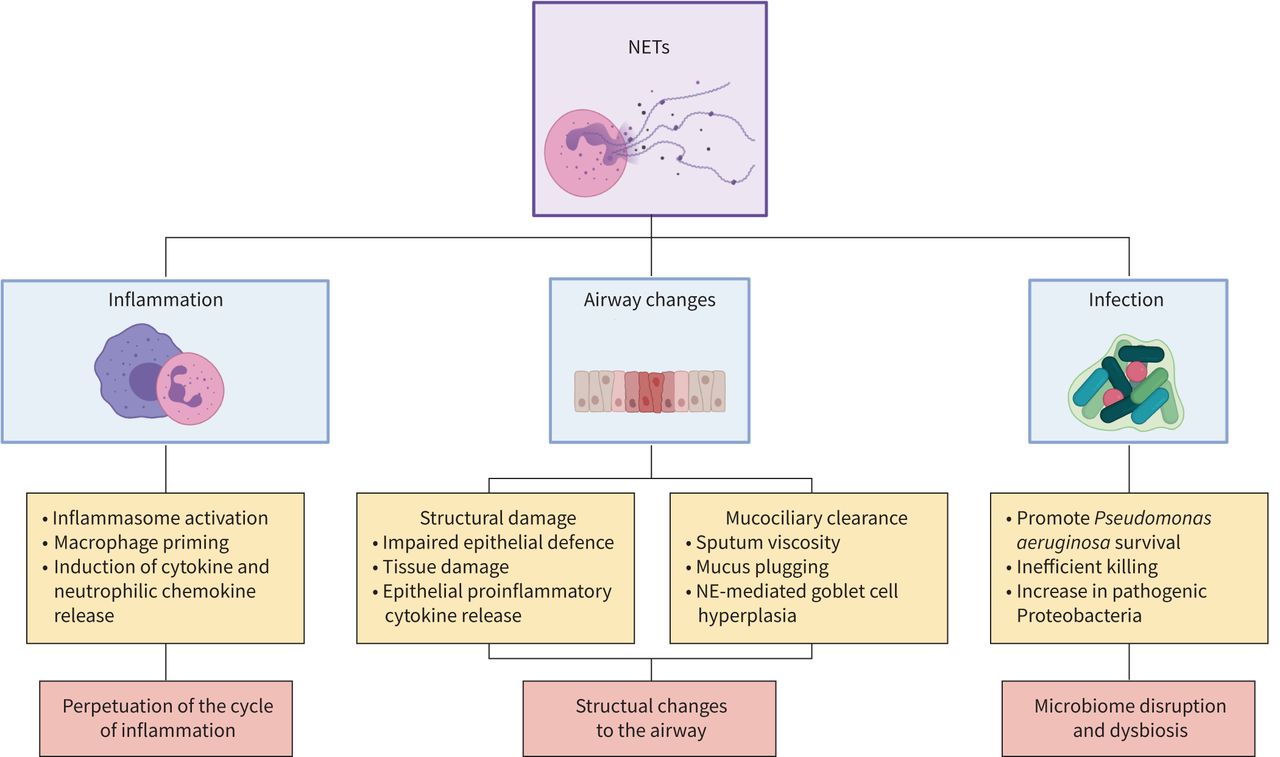{kind=link}
Summary figure of the role of neutrophil extracellular trap (NETs) in chronic lung diseases. Selected effects of NETs on inflammation, mucociliary clearance, lung structure and airway infection are shown. Each are discussed in the text. NE: neutrophil elastase. Figure was created with Biorender.com.
NET-targeting therapies
As neutrophilic inflammation is recognised as part of the pathophysiology of many chronic lung diseases, there have been numerous clinical trials aimed at reducing chronic inflammation. The following sections will give an overview of notable neutrophil-targeting therapies and how these may specifically or non-specifically target NET formation, as summarised in table 1. These therapies are: protease inhibitors, CXCR2 antagonists, DNase treatment, antibiotic and macrolide therapies, and dipeptidyl peptidase 1 (DPP1) inhibition.
Summary of notable neutrophil-targeting therapy studies and clinical trials
Protease inhibitors
It has been well documented that NE causes tissue damage and disruption to the inflammatory response through increasing AHR, goblet cell metaplasia, inflammatory cell accumulation and NET formation. Despite early positive trials in mouse models [75, 82], human clinical trials investigating direct elastase inhibitors have failed to yield positive results. NE inhibitor AZD9668 has been trialled in CF [130], COPD [131] and bronchiectasis [132]. 56 patients were enrolled in the CF trial and randomised to receive either 4 weeks AZD9668 or placebo. Treatment with AZD9668 had no effect on sputum neutrophil counts, NE activity, lung function or clinical outcomes. Similar results were seen in the much larger COPD trial of 838 patients randomised to receive 5 mg twice daily, 20 mg twice daily, 60 mg twice daily or placebo. Treatment had no effect on lung function, respiratory symptoms, quality of life, or lung function. In a trial of patients with bronchiectasis, NE treatment did not reduce neutrophil counts, but did have a small effect on sputum volume. A separate trial published by Watz et al. [133], which trialled the NE inhibitor BAY 85-8501 in a bronchiectasis population, saw no significant changes in lung function or NE activity in sputum. The results of these trials indicate that inflammation is a complex process and that targeting extracellular NE alone may be insufficient.
LTB4
The neutrophil chemoattractant LTB4 has been identified as a potential anti-inflammatory target [64]; however, its effects in larger clinical trials have been mixed. A study of 17 patients with chronic bronchitis and COPD treated with 14 days oral leukotriene synthesis inhibitor produced only very small reductions in some measures of neutrophilic inflammation [134]. In 2014, Konstan et al. [135] published the results of a trial of LTB4 receptor antagonist in children and adults with CF. The trial was designed to recruit 600 patients, but was terminated early after enrolling 155 children and 265 adults due to a significant increase in pulmonary-related serious adverse events (SAEs). In order to understand the increase in SAEs, the authors conduced a follow-up study in a murine model to investigate the effects of LTB4 treatment in models of P. aeruginosa lung infection. The study found that treatment with the LTB4 receptor antagonist reduced pulmonary neutrophil counts and increased P. aeruginosa numbers in the mice lungs. This led to significantly higher levels of bacteraemia and lung inflammation compared placebo-treated mice [136]. The results of this study highlight the risks of anti-inflammatory therapies; although excessive airway inflammation contributes directly to lung damage in patients, decreasing airway neutrophils can lead to more severe infections.
CXCR2 antagonists
Stimulation of the chemokine receptor CXCR2 mediates neutrophil migration into the airways and has been implicated in regulating NET formation via CXCL8 [15]. Clinical trials of CXCR2 treatments in asthma [137], bronchiectasis [138] and CF [139] have shown no clinical benefit for patients, with conflicting results being seen in patients with COPD [67, 140, 141]. After an unsuccessful trial of a competitive and specific CXCR2 antagonist in patients with COPD, we conducted a series of in vitro investigations to characterise the effects of CXCR2 blockade on neutrophil activation. We found that after treating healthy donor cells with COPD sputum, there was no correlation between CXCL8 levels in sputum and subsequent neutrophil activation. This would suggest that there is a subset of patients with COPD in whom neutrophil activation is independent of CXCR2 [67].
This indicates that only a subset of patients would respond to treatment with a CXCR2 antagonist, which may explain why previous studies have failed to yield improved clinical outcomes. The increase in adverse events in both LTB4 and CXCR2 antagonist trials highlights the potentially harmful effects of reducing airway neutrophils. Although airway neutrophilia is established in the pathophysiology of a number of chronic lung diseases, neutrophils are still a vital component of immune defence. Targeted therapies that modulate neutrophil function without compromising recruitment may be beneficial to patients.
DNase treatment
DNA acts as both a structural scaffold and an antimicrobial component of NETs. Furthermore, increased levels eDNA have been linked to disease severity in chronic lung disease, making it a potential therapeutic target. DNase treatment has been trialled with some success, reducing exacerbations and improving pulmonary function, in patients with CF, where patients suffer from extreme mucus plugging due in part to eDNA increasing sputum viscosity [142, 143]. However, trials of DNase in bronchiectasis have been ineffective and in one case harmful to patients [144, 145]. It is possible that the harmful effects of DNase treatment in bronchiectasis is due to NET breakdown and protease release. Dubois et al. [146] published work showing that DNase treatment led to an increase in proteolytic activity of NE, protease 3 and cathepsin G in neutrophils that had been stimulated with P. aeruginosa and S. aureus to release NETs. This suggests that NET-bound NE has reduced cytotoxic properties which are amplified after NET breakdown. This work may explain why DNase treatment was not successful in bronchiectasis, where the release of proteases such as NE actively contributes to tissue damage and inflammation. As CF patients suffer more acutely from mucus plugging compared to non-CF bronchiectasis patients, it is possible that the beneficial effects of reducing sputum viscosity through DNase treatment outweighs the harmful effects of protease release.
Macrolide therapy
Although traditionally known for their antimicrobial properties, there is emerging evidence that macrolide therapies can also have immunomodulatory effects. In 2018, Zimmermann et al. [147] conducted a systematic review of the immunomodulatory effects of macrolides across 1834 participants from 56 randomised controlled trials, prospective cohort studies and case-control studies. They identified that the most reported outcomes were a reduction in neutrophils, reduced concentrations of NE, CXCL-8, IL-6, IL-1β, TNF-α, eosinophilic cationic protein and MMP-9. Several studies have also demonstrated that macrolide treatment can supress NET formation. Azithromycin reduced NET formation through a dose-dependent effect of respiratory burst in neutrophils [148]. Mice exposed to cigarette smoke before treatment with erythromycin had significantly lower levels of eDNA, NE, MPO and citrullinated histone in their BAL fluid than mice receiving a placebo [149]. In a multicentre study investigating the role of NETs in chronic lung disease we analysed sputum NET levels in two cohorts, one in bronchiectasis and one in asthma, of patients receiving 12-month azithromycin treatment. Macrolide treatment in P. aeruginosa-infected patients with bronchiectasis resulted in a statistically significant reduction in NETs compared to subjects not receiving macrolide treatment. Additionally, patients in the AMAZES trial of 12-month azithromycin treatment in asthma had significantly reduced sputum NETs, which was greatest in patients with non-eosinophilic disease at baseline, compared to patients receiving a placebo [94]. Further studies investigating the immunomodulatory effects of macrolide treatment may lead to additional treatments for chronic inflammation, without compromising immune cell function.
DPP1 inhibition
During the neutrophil maturation cycle, activation of neutrophil serine proteases is regulated by the lysosomal cysteine protease DPP1 in the bone marrow. The release of serine proteases by neutrophils can facilitate chronic inflammation and tissue damage, making DPP1 inhibition a potential therapeutic target. Intracellular NE plays a key role in NET formation, suggesting that DPP1 inhibition which blocks NE activation in the bone marrow may reduce NETosis. Papillon–Lefèvre syndrome (PLS) is a rare genetic condition that inactivates cysteine protease cathepsin C, which regulates a variety of serine proteases including NE. Neutrophils from PLS patients are unable to form NETs in response to ROS [150], indicating that DPP1 inhibition may reduce NET formation. A study by Akk et al. [151] found that DPP1 inhibition reduced Sendai virus-induced NET formation in vivo and in vitro. Importantly, PLS patients do not appear to have impaired bacterial killing, suggesting that an approach targeting DPP1 may reduce NETs and inflammation without increasing the risk of infections. A randomised, placebo-controlled study of the DPP1 inhibitor brensocatib (also called AZD7896 or Ins1007) in healthy male volunteers showed that treatment reduced whole-blood NE activity in an exposure-dependent, indirect manner [152]. We recently published the results of a phase 2 randomised controlled trial of brensocatib, in patients with bronchiectasis. 256 patients were enrolled in the study and randomised into three groups: placebo, brensocatib 10 mg and brensocatib 25 mg. Brensocatib treatment significantly increased the time to first exacerbation and reduced the overall number of exacerbations in both treatment groups. Sputum NE concentrations were reduced following brensocatib treatment. Interestingly, patients whose sputum NE remained below the limit of quantification post baseline had a lower incidence of exacerbations [153]. Although these trials are in early phases, these studies indicate that DPP1 inhibition may represent a novel non-antibiotic treatment for patients with chronic lung disease. The main concerns with regard to DPP1 inhibition are related to safety and whether patients may develop features of PLS such as skin hyperkeratosis or dental effects. While the overall safety in the bronchiectasis trial were reassuring, a phase 1 study of a different compound, an irreversible DPP1 inhibitor, found a high frequency of epithelial desquamation, illustrating again the delicate balance between benefit and safety in clinical development [154].
Conclusion
Neutrophilic inflammation and NET formation play a key role in the pathophysiology of asthma, COPD, CF and bronchiectasis. Although NETs do have antimicrobial activity, NET-mediated killing is less effective than phagocytosis and can be utilised by certain pathogens to evade host defence. NET components disrupt the immune response through increasing levels of inflammatory mediators and triggering further neutrophil recruitment to the lungs, thereby intensifying the cycle of inflammation. Local and systemic damage to lung tissue, a hallmark of chronic lung disease, can be exacerbated by NET formation. Past clinical trials aimed at dampening the immune response had mixed success and were accompanied by an increase in adverse effects for some patient populations, highlighting the dangers of reducing neutrophil function in patients with chronic infections. Immunomodulating therapies such as macrolide treatments and DPP1 inhibition may offer NET-targeting treatments that reduce inflammatory mediators without compromising bacterial clearance, offering a new treatment path for patients.
Footnotes
Provenance: Submitted article, peer reviewed.
Conflict of interest: H.R. Keir has nothing to disclose.
Conflict of interest: J.D. Chalmers has received grants or contracts from AstraZeneca, Gilead Sciences, Boehringer Ingelheim, Novartis, Insmed and GlaxoSmithKline; consulting fees from AstraZeneca, Janssen, Boehringer Ingelheim, GlaxoSmithKline, Chiesi, Novartis, Insmed and Zambon; and payment or honoraria for lectures, presentations, speakers’ bureaus, manuscript writing or educational events from Boehringer Ingelheim.
- Received October 22, 2021.
- Accepted December 28, 2021.
- Copyright ©The authors 2022
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References










