





Abstract
The presence of clinical, serological and/or radiological features suggestive, but not confirmatory, of a defined connective tissue disease in patients with interstitial lung disease is a relatively frequent occurrence. In 2015, the European Respiratory Society and the American Thoracic Society proposed classification criteria for the interstitial pneumonia with autoimmune features (IPAF) research entity to capture such patients in a standardised manner, with the intention of nurturing clinical research. This initiative resulted in the publication of several series of IPAF patients, with significant variation between cohorts in clinical characteristics, outcome and the application of IPAF criteria in patient selection. From this increasing body of published work, it has become apparent that revision of IPAF criteria is now required in order to justify the eventual designation of IPAF as a standalone diagnostic term, as opposed to a provisional entity put forward as a basis for clinical research. This review covers the current state of IPAF, conclusions that can and cannot be drawn from the IPAF evidence base, and ongoing uncertainties that require further expert group consideration.
Abstract
Since the proposal of the research entity interstitial pneumonia with autoimmune features (IPAF) in 2015, many questions remain unanswered and challenges remain in its definition https://bit.ly/38GDbLE
Introduction
The diffuse parenchymal lung diseases are differentiated from the outset by the presence of an established cause or association. Connective tissue diseases (CTDs) represent one of the more common causes of non-idiopathic interstitial lung disease (ILD). Almost all CTDs may be associated with ILD, but the individual CTDs vary in the prevalence and most frequent morphological pattern of ILD. CTDs are defined by strict diagnostic criteria that, outside of systemic sclerosis (SSc), exclude ILD as a diagnostic factor. When ILD complicates a previously defined CTD, the diagnosis is generally straightforward, amounting to CTD-ILD (with the exception of drug-induced disease). However, a reasonable proportion of patients present with ILD in the presence of features suggestive of, but not confirmatory for, a defined CTD. Whether such patients behave and respond similarly to CTD-ILD, or more akin to their idiopathic counterparts, has been the focus of much literature over the last decade. Despite defined classification criteria for research under the title interstitial pneumonia with autoimmune features (IPAF), challenges and controversies continue to surround this group of patients.
Why was IPAF proposed?
A sizable proportion of patients with ILD present with clinical, serological and/or radiological features suggestive of CTD, but lacking features to meet the established diagnostic criteria of defined CTDs [1–7]. Up to 2015, this overlap between idiopathic interstitial pneumonia (IIP), especially idiopathic pulmonary fibrosis (IPF) and CTD-ILDs (figure 1) was described using a variety of terminologies, including “undifferentiated CTD-ILD” [8], “undifferentiated connective tissue syndromes” [9, 10], “lung-dominant CTD” [11], “early CTD”, “formes frustes” of CTD [1], “occult CTD”, “overlap CTD”, “lung-limited CTD” [2] and “autoimmune-featured ILD” [12]. These terms largely overlap yet do not exactly match. Corresponding studies provided accumulating evidence that CTD features were relevant in the setting of ILD even when falling short of fulfilling existing classification criteria for a specific CTD. However, research was hampered by heterogeneity in terminology and definition [13].

Interstitial pneumonia with autoimmune features (IPAF) in the schema of clinical interstitial lung disease (ILD).
In 2015, the European Respiratory Society (ERS) and American Thoracic Society (ATS) Task Force on Undifferentiated Forms of Connective Tissue Disease-Associated Interstitial Lung Disease proposed classification criteria for a so-called research category IPAF [14], with the aim of providing a consensus terminology and definition, and to foster research. The Task Force included respiratory physicians, rheumatologists and immunologists. The IPAF classification criteria are summarised in table 1.
Classification criteria for interstitial pneumonia with autoimmune features
Advantages of the IPAF concept and definition
The concept of IPAF emphasises that there can be an overlap between IIPs and CTD-ILD and provides a framework to designate and study this condition. Such overlap between entities is common in medicine, and can be due to overlapping manifestations, as seen in overlap CTD, in which features of separate CTDs are present in the same patient, or be related to uncertain aetiology, as in patients with a condition strongly suggesting IPF but exposed to inhaled antigens known to cause chronic fibrotic hypersensitivity pneumonitis [15]. IPAF deals with the overlap between IIPs and CTD-ILD.
The concept of IPAF has highlighted the need for multidisciplinary discussions in the field of CTD-ILD, especially the input of rheumatologists in the ILD clinic [16]. It has already shed light on the importance of a thorough evaluation of patients with apparently idiopathic ILD and on the value of the interaction between medical specialties [17]. Although its management is not standardised, the concept of IPAF facilitates making decisions in patients presenting with ILD and mild CTD features before a longer follow-up clarifies whether CTD is present or not [8], as opposed to the concept of “undifferentiated CTD”, therefore addressing an unmet clinical need when seeing a patient with new-onset ILD.
Outstanding issues in the IPAF concept
A question that remains open is whether IPAF represents a clinical diagnosis, a distinct entity, or just an overlap of existing conditions. A small proportion (10–20%) of patients initially classified as IPAF progress to a diagnosis of CTD-ILD [18–22], suggesting that in some cases IPAF does indeed correspond to CTD-ILD early in the course of disease. Also, significant heterogeneity persists within the group designated by IPAF. The respective contribution of the different domains to the classification as IPAF varies between series [23], suggesting variations in referral patterns, with some IPAF series being “close to IPF cases” and others “closer to CTD-ILD cases”. The outstanding issues surrounding the IPAF criteria are detailed below and summarised in table 2.
Unresolved issues of current interstitial pneumonia with autoimmune features (IPAF) criteria
Inclusion of rheumatological review
Routine involvement of a rheumatologist in the multi-disciplinary evaluation of patients with ILD has been proposed but is not a current prerequisite for the adjudication of IPAF criteria. Chartrand et al. [24] and Jee et al. [25], whose clinics routinely included rheumatologists, noted a higher prevalence of features from the clinical domain. Additionally, the IPAF cohort of Ahmad et al. [26] was notable for a number of patients with abnormal nailfold capillaroscopy and salivary gland biopsy, investigations not typical of a pulmonologist. In fact, abnormal nailfold capillaroscopy, together with the presence of ILD, may be sufficient to satisfy SSc diagnostic criteria (table 3), removing such cases from the IPAF entity [27]. Levi et al. [28] observed that, following rheumatological review, 40% of selected ILD multi-disciplinary meeting cases received a rheumatology-related ILD diagnosis, including IPAF. One-fifth of IPF diagnoses were reclassified into a range of CTD-ILDs. However, the data supplement would suggest that pulmonologists had access to a narrow range of auto-antibodies, and it is unclear whether defined CTD criteria were enforced. De Lorenzis et al. [29] evaluated the routine involvement of rheumatological expertise in their cases with a suggestion of an underlying rheumatic disorder. Interestingly, a large number of patients underwent additional investigations, including articular radiology, electromyography, nailfold capillaroscopy and salivary gland biopsy. Agreement between pulmonologists and rheumatologists across various CTD clinical and serological features was only fair to moderate, with rheumatologists identifying a greater number of extrapulmonary features of CTD. To a large extent, identifying IPAF criteria depends on the effort made to look for CTD features. Nailfold capillaroscopy [26], biopsy of accessory salivary glands [26, 30, 31] or consultation with a rheumatologist or a dermatologist often identify CTD features that may alter the eventual diagnosis, yet they cannot be easily recommended in all patients with ILD. The decision on how far to search for evidence of an underlying CTD often impacts the eventual diagnosis [32]. The current IPAF criteria do not specify a need for specialist rheumatologist evaluation, but the data summarised above suggest that rheumatological input might help to improve the sensitivity and specificity of the IPAF criteria [33].
Various diagnostic criteria proposed for interstitial lung disease (ILD) in patients with features of connective tissue disease (CTD), and the diagnostic criteria for defined CTDs adapted to suit the domains of the interstitial pneumonia with autoimmune features (IPAF) criteria
Controversies surrounding inclusion/exclusion of items/characteristics
The heterogeneity in IPAF series has prompted some authors to suggest alternative definitions of IPAF [34, 35], to remove some of the individual items from the IPAF group, especially anti-synthetase antibodies [36, 37] (see below), to consider the inclusion of selected additional items (e.g. sicca symptoms, oesophageal dysmotility, lymphocytic bronchiolitis on biopsy, proximal muscle weakness with myalgia [38] or antineutrophil cytoplasmic antibodies [39]), or to separate subjects with a usual interstitial pneumonia (UIP) pattern from those with a non-UIP pattern (figures 2 and 3) [35].
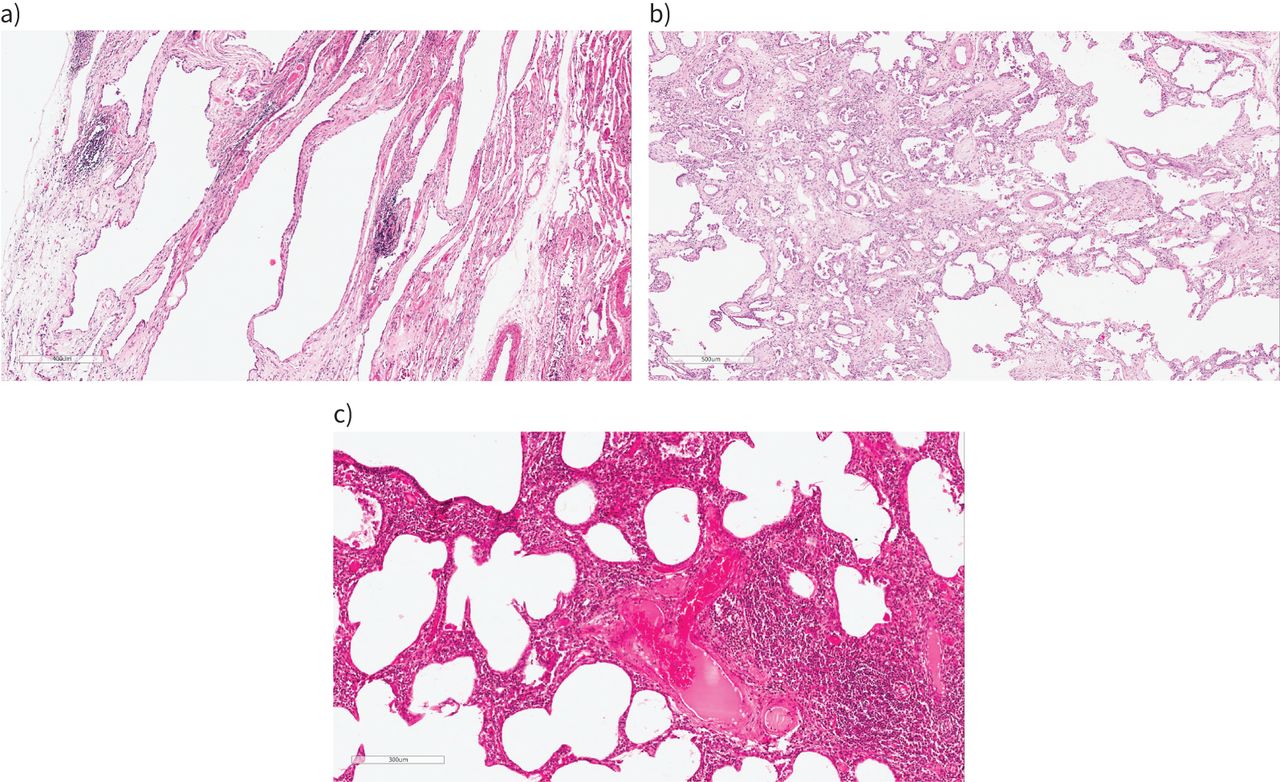
Histological patterns of interstitial pneumonia seen in interstitial pneumonia with autoimmune features. a) Fibrotic non-specific pneumonia (NSIP): there is diffuse uniform interstitial fibrosis associated with a mild patchy non-specific chronic inflammatory cell infiltrate. b) Organising pneumonia (right side) with progression to interstitial fibrosis (left side): buds of granulation tissue merge with established fibrosis. c) Lymphoid interstitial pneumonia (LIP) overlapping with cellular NSIP: in this field, the right-hand part of the image shows a density of interstitial inflammation characteristic of LIP.

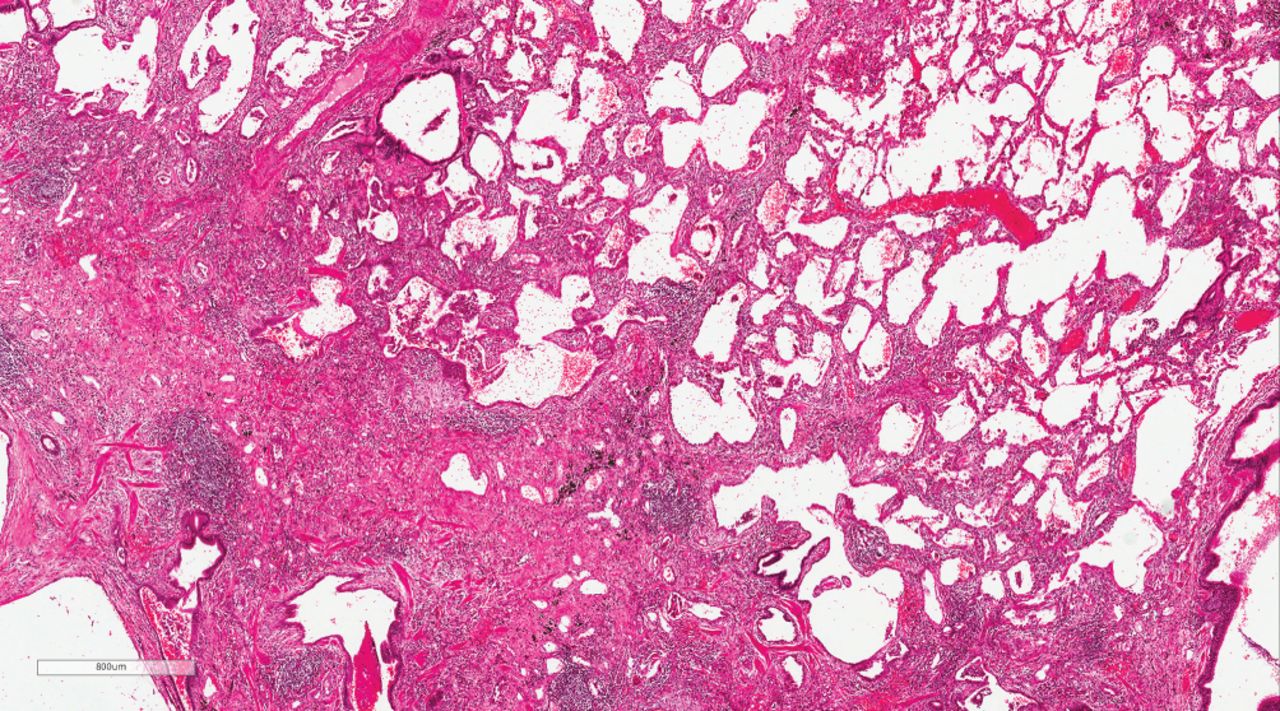
Usual interstitial pneumonia (UIP) with features arguing against idiopathic pulmonary fibrosis (IPF). There are areas with established patchy interstitial fibrosis with an occasional fibroblastic focus characteristic of UIP, but also areas where interstitial chronic inflammation predominates, making this case indeterminate for UIP/IPF. Based on pathology alone, either fibrotic hypersensitivity pneumonitis or a connective tissue disease-related interstitial lung disease (interstitial pneumonia with autoimmune features) were favoured over IPF, and multidisciplinary review was recommended.
The issue of sicca symptom inclusion is an example of some of the uncertainties surrounding the inclusion of specific items in the clinical domain. In reports prior to the IPAF nomenclature, sicca symptoms had been invariably included as an autoimmune clinical feature. However, sicca symptoms are absent from IPAF criteria, as they are considered to lack specificity. While Corte et al. [10] and Kinder et al. [9] showed no difference in the prevalence of sicca symptoms between those with and without autoimmune features, Vij et al. [12] reported a prevalence of 57.1% compared to 15.5% in those with autoimmune features and IPF respectively. Sebastiani et al. [22] observed a similar prevalence of sicca symptoms in their IPAF cohort, suggesting that their inclusion may need to be considered.
Myositis-specific antibodies (MSAs), including anti-synthetase antibodies, are an example of the uncertainties surrounding the serological domain (figure 4a and b). MSAs are currently included in the IPAF serological domain (table 1). Graham et al. [37] compared survival within IPAF patients, subdivided according to whether patients were 1) positive for MSAs; 2) positive for myositis-associated antibodies (MAAs) but negative for MSAs; or 3) negative for both. The study also included cohorts of idiopathic inflammatory myositis (IIM), CTD-ILD and IPF patients. Interestingly, IPAF patients with MSA (n=35), but not MAAs (n=65), were more frequently characterised by a computed tomography (CT) or pathology pattern of non-specific interstitial pneumonia (NSIP), and had very good outcomes on immunosuppressive treatment, similarly to patients with IIM, and significantly better than IPAF with MAA, or IPAF without MSA/MAA. While a UIP pattern was associated with a worse survival in IPAF overall, and in the IPAF-MAA group, this was not the case in IPAF-MSA, although there were only six cases with IPAF-MSA UIP. Whether a UIP pattern associated with MSA has a better prognosis than the other IPAF UIP patients will require confirmation in further studies. Huapaya et al. [40] reported progressive improvement and/or stabilisation with azathioprine or mycophenolate in patients with IIM. Graham et al. [37] suggested that IPAF with MSA should be clinically managed as IIM ILD, and that MSA should potentially be removed from the IPAF criteria.

a and b) Interstitial pneumonia with autoimmune features (IPAF) in a 60-year-old male presenting with acute onset type 1 respiratory failure. Computed tomography (CT) demonstrates patchy consolidation with a bronchocentric distribution, and some lower lobe volume loss, suggestive of a pattern of fibrotic organising pneumonia. Positive serology for anti-OJ was identified. No clinical features of an idiopathic inflammatory myopathy were evident at presentation. The patient had an excellent response to prednisolone and tacrolimus. c and d) IPAF in an 80-year-old male. The CT demonstrates a usual interstitial pneumonia (UIP) pattern with right-sided pleural thickening/effusion suggestive of multi-compartment involvement. No known history of asbestos exposure was reported and no pleural plaques were evident on CT. The patient's anti-nuclear antibody (ANA) was low titre but in a nucleolar pattern and rheumatoid factor was elevated. The patient was treated with immunomodulation, but demonstrated an idiopathic pulmonary fibrosis-like disease trajectory and died within a few years of diagnosis. e) 84-year-old female presenting with gradually progressive dyspnoea with CT demonstrating a UIP pattern. ANA at presentation was speckled with titre of 1:320. The patient subsequently developed inflammatory arthritis and a positive anti-cyclic citrullinated peptide, resulting in an eventual diagnosis of rheumatoid arthritis and associated interstitial lung disease. Her rheumatoid arthritis was treated with sulfasalazine and the lung disease has remained under observation after the patient declined clinical trial participation. Over the first 12 months there was a 7% relative decline in forced vital capacity and a 26% relative decline in carbon monoxide diffusing capacity.
Clustering in time of the manifestations (e.g. concomitant apparition of arthralgia and gastro-oesophageal reflux in a patient with new-onset ILD) may be meaningful clinically, but it is not captured in the IPAF criteria. Identification of a family history of autoimmune disease by Chartrand et al. [24] in 25% of their IPAF cohort suggests that this factor might also require consideration.
The evaluation of multi-compartment involvement
The IPAF morphological domain requires either the presence of a non-UIP histological pattern or “multi-compartment involvement”, i.e. co-existing airways disease, pulmonary vascular disease or pleural disease (figure 4c and d). However, there is a lack of specificity and reproducibility of the multi-compartment involvement subdomain as an independent criterion. There are ongoing difficulties with the identification of co-existent disease processes relevant to the entity of IPAF due to the high prevalence of comorbidities in patients with ILD.
In this regard, the definition of “disproportionate” pulmonary vascular disease, a well-recognised autoimmune phenotype, is especially problematic. In the index IPAF series of Oldham et al. [41], pulmonary vascular involvement was identified based on a reduction in carbon monoxide diffusing capacity (DLCO) that was disproportionate to forced vital capacity (FVC), captured using an FVC/DLCO threshold. However, whilst standardisation of this sort has advantages, an FVC/DLCO threshold is not, in itself, indicative of disproportionate pulmonary vasculopathy. Pulmonary hypertension is a frequent complication in advanced fibrotic lung disease and, when severe, is associated with major increases in the FVC/DLCO ratio and major reductions in the gas transfer coefficient (KCO). However, pulmonary hypertension is not a disproportionate finding in this scenario and is not an “autoimmune feature”. Moreover, both KCO and the FVC/DLCO ratio are non-specific: influenced alike by pulmonary vasculopathy and the presence of concurrent emphysema. In the “idiopathic” (or tobacco-related) combined pulmonary fibrosis and emphysema syndrome, and in patients with combined emphysema and CTD-ILD, DLCO levels are disproportionately reduced whilst lung volumes, including FVC, tend to be spuriously preserved. Thus, the use of standardised FVC/DLCO or KCO thresholds to define multi-compartment disease may have led to false positive satisfaction of IPAF criteria in some patients. Indeed, this problem may lead to the selective designation of IPAF in patients with advanced ILD or concurrent emphysema, confounding outcome comparisons between patients meeting IPAF criteria and those with IPF or idiopathic NSIP.
The identification of airway disease also poses difficulties, whether identified by ventilatory patterns or CT findings. It is often difficult to decipher whether the presence of airway disease is in favour of an underlying CTD, what exactly constitutes airway disease in subjects with fibrotic ILD, and how the confounding effect of tobacco smoking can be dealt with rigorously. An obstructive or mixed ventilatory defect may result from concurrent asthma or smoking-related chronic obstructive pulmonary disease. Furthermore, no pulmonary function threshold has been validated in the identification of airway disease when there is concurrent ILD. The presence of “standalone bronchiectasis” on CT in lung regions distant from interstitial fibrosis (as seen in rheumatoid arthritis (RA) or Sjögren disease, but quite rarely in IPF) is likely to be a robust multi-compartment criterion. However, the distinction between standalone bronchiectasis and traction bronchiectasis is not always straightforward, especially when ILD is extensive.
Thus, multi-compartment disease, when obviously present, is an important autoimmune feature but its definition will need to be carefully considered when IPAF criteria are eventually revised. We favour a robust approach in which pulmonary vasculopathy is considered to be disproportionate only when ILD is mild in severity (whether defined by CT or pulmonary function variables) and emphysema is absent or limited in extent. There is no current consensus on pulmonary function or CT extent thresholds that might be harnessed for this purpose. This will require careful expert group consideration of whether current pulmonary hypertension criteria should be discarded from the designation of multi-compartment disease in IPAF or modified to specify disproportionate pulmonary vasculopathy. Similarly, it can be argued that airway disease should be designated only when there is standalone bronchiectasis on CT or an unexplained obstructive or mixed ventilatory defect (i.e. in the absence of asthma or smoking-related emphysema).
Prognostic implications of the IPAF entity
Ultimately, the introduction of a new entity is useful if it provides prognostic and/or management implications. In the article by Oldham et al. [41] that quickly followed the IPAF proposal, approximately a third of patients with an IIP or “undifferentiated” CTD met IPAF criteria, including 18% of their IPF cohort. There have since been numerous studies detailing the features and outcomes for various IPAF cohorts [20, 24–26, 42–48]. A summary and comparison of published IPAF cohorts can be found in table 4 [25]. However, despite the ERS/ATS IPAF research statement, significant cohort heterogeneity remains, resulting at least in part from the retrospective nature of most series and of varying recruitment patterns by centre. The selection of subjects to whom IPAF criteria are applied across studies has differed, as has the application of the multi-compartment morphological domain criterion. Some of the inconsistency in reporting relates to the presence or absence of certain autoantibodies in the systematic screening of patients with IIP. As a result, the various IPAF studies report on slightly different patients and their associations with outcomes.
Comparison of interstitial pneumonia with autoimmune features (IPAF) cohorts
In the cohort of Oldham et al. [41], subjects most commonly met IPAF criteria on the basis of combined serological and morphological features. Supporting the validity of the IPAF criteria was the observation that survival trended approximately halfway between that of true CTD-ILD and IPF, an observation that has been noted variably in other studies. In the study by Oldham et al. [41], once the presence of a UIP pattern was taken into account, survival of UIP-IPAF was identical to IPF, while non-UIP IPAF had outcomes closer to those of CTD-ILD, suggesting that IPAF features in the context of a UIP pattern may be of limited prognostic relevance. However, a subsequent study from the same centre, while identifying honeycombing as being independently associated with adverse survival, did not identify a clear survival difference between definite and inconsistent for UIP radiological patterns in IPAF subjects [49]. A recent systematic review and meta-analysis confirmed the poor outcome associated with a UIP pattern in the context of IPAF [50]. The review acknowledged additional recognised risk factors for IIP mortality in IPAF subjects, including age, male gender, smoking history and lung function parameters. However, on multivariable analysis, older age was the only parameter significantly associated with mortality [50].
Should a UIP pattern be excluded from the IPAF entity?
In IPAF, a UIP pattern is excluded from the morphological domain as an IPAF criterion. However, if additional features suggestive of autoimmune or multi-compartment involvement are observed, including diffuse lymphoplasmacytic infiltration, interstitial lymphoid aggregates, histological involvement of the airways, unexplained pleural disease and unexplained pulmonary vasculopathy, the IPAF morphological domain can be met even with a UIP pattern (table 1) (figure 4c and d). Furthermore, a patient with a UIP pattern and no additional morphological features can be included within IPAF if at least one item in each of the clinical and serological domains is present (table 1). In CTD-ILD, a UIP pattern is generally associated with a better survival than UIP/IPF [51–53]. RA may be an exception, as RA-ILD patients with a definite and extensive UIP pattern on CT and/or a UIP pattern on biopsy have an IPF-like survival [54–58]. The fact that, in most studies, a UIP pattern in the context of CTD-ILD is not associated with an IPF-like behaviour (with the exception of RA-ILD), supported not excluding patients with UIP (providing they met IPAF criteria) from the IPAF entity. However, in light of the poor prognostic impact of a UIP pattern within IPAF, there could be an argument for its removal. Nevertheless, there are a number of issues that will need to be considered before deciding whether IPAF-UIP needs to be considered separately [35]. Not all IPAF-UIPs are equal and demographic/clinical characteristics will impact on prognostic and management implications. Furthermore, it is possible that UIP-IPAF would not have the same outcome as IPF if the IPAF criteria were revised, with a particular focus on allowing satisfaction of the multi-compartment criterion through vasculopathy, just because an IPF patient has PH. In addition, the exact definition of increased inflammation in the context of a UIP biopsy, and whether more intense/numerous diffuse lymphoplasmacytic infiltration and/or interstitial lymphoid aggregates are associated with a better prognosis, remains unclear. Figure 3 is an example of histological UIP with areas of interstitial chronic inflammation.
How does the presence of autoimmune features influence initial evaluation?
At present, no consensus exists on best practice with regard to the investigation and initial management of patients meeting IPAF criteria. We stress that the views expressed below are the combined views of the authors.
According to studies published so far, a UIP pattern in the context of IPAF, whether on CT or at biopsy, is associated with a similar mortality to IPF [41, 44]. In the appropriate clinical context for an IPF diagnosis (age greater than 60 years, male sex and a smoking history), we believe that a probable UIP pattern on CT, even in patients meeting IPAF criteria, may not require histological confirmation of UIP, as management would not necessarily change. In contrast, the threshold for biopsy in the context of a probable UIP pattern on CT is reduced in the younger IPAF patient, particularly if female (see treatment section below). In most studies, although not in all [26], patients with non-UIP IPAF have a similar survival to CTD-ILD, and most ILD specialists would probably treat in a similar manner, at least initially. In our opinion, while identification of a histological NSIP pattern would support an immunosuppressive approach, histological confirmation of a UIP pattern would lead to a more cautious approach to immunosuppression, with frequent monitoring and earlier consideration of antifibrotic therapy, should there be progression. Finally, the finding of associated morphological features of IPAF on a biopsy otherwise characterised by a UIP pattern, would lead to further interrogation and monitoring over time to detect additional features suggestive of an underlying CTD. Whether cryobiopsy samples, rather than those obtained through a surgical biopsy, are sufficient to detect these additional features in the context of IPAF has not been established, and the decision as to which biopsy procedure to undertake and whether to proceed to surgical biopsy if less invasive techniques are inconclusive needs to be made on a case-by-case basis. In light of the many complexities in defining IPAF criteria, these cases will always benefit from discussion by a multidisciplinary team, with input from pulmonologists, radiologists, pathologists, rheumatologists and, in some instances, immunologists.
Monitoring of autoimmune features over time
ILD may be the initial manifestation of a CTD, and it is therefore possible for patients with ILD, including IPAF, to manifest a defined CTD at a later timepoint (figure 4e) [59]. Sebastiani et al. [22] observed IPAF evolution to definite CTD in 13.5% of cases. A definite CTD diagnosis of Sjögren disease, RA and polymyositis was possible after a mean of 31 months (range 7–71 months). In two cases, these diagnoses were made after the development of relevant antibody positivity which was absent at baseline. In a cohort of patients with an initial diagnosis of idiopathic NSIP, 10% developed clinical manifestations of a defined CTD at a median of almost 2 years after their ILD diagnosis [60]. It is important to recognise that IPAF is not a prerequisite for the subsequent development of a CTD.
All patients with an ILD should be routinely evaluated for the presence of CTD symptoms and signs, with re-evaluation occurring at each visit and for the duration of follow-up, even in the case of a highly confident diagnosis of IPF. In cases of IPAF, the treating clinician should continually search for those features which will add to the CTD diagnostic puzzle. Cases which satisfy IPAF criteria should be considered for formal rheumatological evaluation, as a defined CTD, following established criteria (table 3), will better inform treatment decisions. The goals are to achieve a highly confident diagnosis and to inform a patient of the aetiology, natural history and treatment of their disease. However, lower degrees of diagnostic confidence are often the best that can be achieved at presentation, with the need for an iterative diagnostic approach, and repeated multi-disciplinary discussions as needed, taking into account response to initial treatment, disease behaviour and development of additional features over time. Re-evaluation with repeat anti-nuclear antibody (ANA) testing is unlikely to be informative [61], noting that a positive ANA on its own, even when in an anti-centromere pattern, is not sufficient for systemic lupus erythematosus or SSc. However, repeating extended antibody testing should be considered, particularly in those with radiological/histological phenotypes of NSIP, organising pneumonia (OP) or a combination of both, and/or clinical features suggestive of, but not confirmatory for, SSc or IIM. Involvement of an immunologist may be necessary in such a scenario, in order not to miss the rare autoantibodies that are not systematically assessed. The development of an inflammatory arthralgia in a patient with ILD should herald the potential development of RA, noting that positive serology is not necessary for a diagnosis (figure 4e). Just as important as the search for definite CTD features are the efforts to exclude a CTD, particularly in the case of UIP radiology. Where the IPAF criteria are satisfied only for low titre ANA and perhaps borderline multi-compartment involvement (figure 4c and d), a diagnosis of IPF should be considered, although perhaps with a degree of confidence.
Treatment of IPAF
As IPAF is a research entity and not a diagnosis, studies specifically addressing the management of IPAF are lacking. There have been no randomised controlled trials specifically conducted in IPAF. Management strategies must therefore be based on expert opinion and are largely extrapolated from CTD-ILD studies [17, 62]. It is uncertain whether a specific management distinct from that of IPF and from CTD-ILD is needed; however, research is required to address this question. In the absence of a general approach that could be applied to any patient with lung disease fitting the definition of IPAF, management is individualised.
Similarly to other ILDs, especially IPF [63], pulmonary rehabilitation, long-term oxygen supplementation therapy if appropriate, and treatment of gastro-oesophageal reflux if present, are indicated, as well as prevention of infection and bone health assessment. In advanced disease, lung transplantation should be considered, extrapolating satisfactory outcomes in appropriately selected patients with CTD, including SSc [64].
In the series of IPAF published to date where treatment is reported [23, 25], a majority of patients have received glucocorticoids and/or immunosuppressive therapy, including mycophenolate, azathioprine, cyclophosphamide, calcineurin inhibitors (ciclosporine and tacrolimus) and occasionally rituximab, illustrating that many clinicians would treat subjects with IPAF as they would treat CTD-ILD [65–68]. Figure 5 depicts a case meeting IPAF criteria treated similarly to CTD-ILD with pulsed methylprednisolone and cyclophosphamide, followed by maintenance mycophenolate and prednisolone. A small minority of patients in published series have received antifibrotic agents [25], which may reflect the fact that until recently these drugs were only approved and available for patients with IPF. In these series, however, efficacy of management is virtually impossible to assess in the absence of a control group and given the high clinical heterogeneity.

Interstitial pneumonia with autoimmune features (IPAF) in a 32-year-old male presenting with subacute cough and dyspnoea over 2 months. On presentation, he was in type 1 respiratory failure requiring supplemental oxygen. a) His computed tomography (CT) demonstrated upper zone ground glass opacity and fibrotic organising pneumonia in the lung bases. Anti-nuclear antibody was positive at 1:1280, although extended serology was negative. There were no clinical features of an autoimmune condition. IPAF criteria were met by morphological and serological domain. b) A significant response was observed with immunomodulation, with ongoing lung function improvement over a protracted period. FVC: forced vital capacity; i.v.: intravenous; KCOc: gas transfer coefficient, corrected for haemoglobin; MP: methylprednisolone; o.d.: once daily; TLCOc: transfer factor of the lung for carbon monoxide, corrected for haemoglobin.
There are data stemming from studies of patients with unclassifiable ILD, some of which include patients with IPAF. In one study of patients with unclassifiable ILD, intravenous pulse cyclophosphamide was suggested to stabilise lung function [69]; a subset of patients in this study had IPAF and these seemed to benefit more from the treatment regimen, although none of them had a UIP pattern. This suggests that patients with IPAF and a non-UIP pattern may benefit from immunomodulation, although this needs confirmation.
In a retrospective cohort of 52 patients who met criteria for IPAF, 28 received mycophenolate after a median time of 22 months [70]. Although changes in FVC% and DLCO% were not significantly different between the mycophenolate-treated and untreated groups, there was a trend toward more rapid baseline decline of both FVC% and DLCO% in the mycophenolate-treated cohort before versus after mycophenolate therapy. This suggests that patients with IPAF might benefit from mycophenolate therapy; however, this view is again subject to many possible biases.
In a multicentre, double-blind, randomised, placebo-controlled phase 2 trial, 253 patients who had progressive fibrosing unclassifiable ILD were randomised to receive pirfenidone versus placebo [71]. Randomisation was stratified by concomitant mycophenolate mofetil use and presence or absence of IPAF. Analysis of the primary endpoint, the mean predicted change in FVC from baseline over 24 weeks measured by daily home spirometry, was affected by intraindividual variability, which prevented application of the prespecified statistical model. Over 24 weeks, predicted mean change in FVC measured by site spirometry was lower in patients given pirfenidone than placebo (treatment difference 95.3 mL, 95% CI 35.9–154.6; p=0.002). Compared with the placebo group, patients in the pirfenidone group were less likely to have a decline in FVC of more than 5% or more than 10%. At week 24, mean change in DLCO from baseline was −0.7% (standard deviation 7.1) for the pirfenidone group and −2.5% (8.8) for the placebo group, and mean change in 6-min walk distance from baseline was −2.0 m (68.1) for the pirfenidone group and −26.7 m (79.3) for the placebo group. Changes from baseline in University of California San Diego–Shortness of Breath Questionnaire, Leicester Cough Questionnaire score, cough visual analogue scale and St George's Respiratory Questionnaire scores were similar between the pirfenidone and placebo groups at week 24. The safety and tolerability profile was acceptable. This study suggests that patients with progressive fibrosing unclassifiable ILD could benefit from pirfenidone treatment. Only 33 patients with IPAF were included in this study, precluding any conclusion regarding this subgroup.
Nintedanib, a tyrosine kinase inhibitor with antifibrotic properties, has demonstrated efficacy in IPF (the INPULSIS studies) [72], in SSc-associated ILD (the SENSCIS study) [73] and in patients with ILD and a progressive fibrosing phenotype (the INBUILD study) [74]. In the SENSCIS study, approximately half of the patients were also receiving mycophenolate, demonstrating that nintedanib can be combined with mycophenolate with acceptable tolerance. In the INBUILD study, 114 participants (17%) had unclassifiable ILD; however, the proportion of them who met IPAF criteria is unknown. Sensitivity analysis of the annual rate of decline in FVC (mL·year–1), excluding aetiological subgroups of ILD diagnosis, demonstrated homogeneity of results across subgroups [75]. Although the INBUILD trial was not designed or powered to provide evidence for a benefit of nintedanib in any specific diagnostic subgroup, the results suggest that nintedanib reduces FVC decline in patients who have a chronic fibrosing ILD and progressive phenotype, irrespective of the underlying ILD diagnosis. It can be inferred that subjects with IPAF who have a progressive phenotype can benefit from nintedanib treatment.
Overall, possible treatment decisions in patients classified as with IPAF include observation without drug therapy, immunomodulation with glucocorticoids and/or immunosuppressants, antifibrotic drugs, and a combination (upfront or sequential) of immunomodulation and antifibrotics. Currently, treatment decisions must be based on a careful evaluation of benefit to risk ratio in the individual subject and should be discussed in a multidisciplinary setting. Eventually, the choice of first-line therapy is based on the global assessment of the patient, including the age and gender of the patient, the type of extrapulmonary manifestations present, the underlying CTD for which there is a “flavour”, the chest CT pattern and the histopathological pattern if available, the severity of functional impairment, the existence of disease progression, the patient's expectations, the comorbidities and co-medications, and the time elapsed since the diagnosis of ILD (figure 6). Of those, the demographics and the imaging phenotype are often the main elements to take into consideration, separating a predominantly fibrosing from a predominantly inflammatory phenotype.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Factors to consider in the management of patients fulfilling interstitial pneumonia with autoimmune features criteria. CTD: connective tissue disease; CT: computed tomography.
As an example, a 65-year-old male patient with a chest CT pattern of NSIP and a UIP pattern on biopsy might not benefit from corticosteroids and/or immunosuppressive drugs even if lymphocytic follicles are present on biopsy and high titre of antinuclear antibodies with nucleolar pattern are found; a sensible decision in such case may be to observe and monitor disease progression, and later to treat using antifibrotics, in case the clinical behaviour is that of a progressive phenotype. Conversely, a 50-year-old female with a chest CT suggesting NSIP and a high titre of antinuclear antibodies with nucleolar pattern, who declined lung biopsy, will likely be treated similarly to SSc-associated ILD even in the absence of any other manifestation of CTD. Similarly, a chest CT pattern of OP associated with NSIP is highly suggestive of autoimmune myositis and is likely to be managed as such by most ILD experts, even in the absence of autoantibodies identified precluding the classification of such cases as autoimmune myositis.
Next to these extreme examples, many cases fall into a spectrum that encompasses both inflammatory/autoimmune and fibrotic features, hence presenting very challenging management decisions. In the future, it is likely that a combination of approaches [76] may be used in many patients with IPAF. One key element to keep in mind is the detrimental effect of high-dose glucocorticoids and azathioprine therapy in patients with IPF, potentially applicable also to IPAF patients with UIP. Furthermore, as IPAF is not a validated as a standalone diagnosis, our view is that in most cases we should, for management purposes, view IPAF-UIP as synonymous with IPF. The designation of IPAF as a diagnosis deprives these patients of access to antifibrotic therapy at presentation. This is a particular problem if pulmonary vasculopathy is viewed as a separate process in IPF and is the basis for the designation of multi-compartment disease. As discussed earlier, Oldham et al. [41] classified UIP patients as having IPAF based on an inflexible FVC/DLCO ratio and one other criterion. In the majority of cases, the existing validated diagnosis in patients with IPAF-UIP is IPF, with the crucial consequence that these patients are eligible for antifibrotic therapy at presentation.
Progress in management is critically dependent on randomised clinical trials and of prospective registry studies dedicated specifically to patients with IPAF, with the aim of guiding individualised medicine based on better phenotyping and more precise characterisation of all features present.
Conclusion
The authors who collaborated on the ERS/ATS IPAF research statement indicated upfront that this represented a “first draft”, to be considered as a starting point for future work. It is not surprising, therefore, that numerous suggestions have been proposed to improve and refine the IPAF criteria. There remains an important ongoing need for the IPAF entity, serving a valuable purpose in identifying those cases with autoimmune features that do not meet criteria for defined CTDs. Much more is known about these cases than in 2015 when IPAF was proposed, and it might now be time to work on a “second draft” of the ERS/ATS IPAF statement.
Acknowledgements
Mouhamad Nasser (National Reference Center for Rare Pulmonary Diseases, Louis Pradel Hospital, Lyon, France) contributed towards the production of this manuscript.
Footnotes
Provenance: Commissioned article, peer reviewed.
Conflict of interest: J.A. Mackintosh has nothing to disclose.
Conflict of interest: A.U. Wells reports personal fees from Roche, Boehringer Ingelheim and Bayer outside the submitted work.
Conflict of interest: V. Cottin reports personal fees and non-financial support from Actelion; grants, personal fees and non-financial support from Boehringer Ingelheim; personal fees from Bayer/MSD; personal fees and non-financial support from Roche/Promedior; and personal fees from Sanofi, Celgene/BMS, Galapagos, Galecto, Shionogi, AstraZeneca, Fibrogen, RedX and PureTech, outside the submitted work.
Conflict of interest: A.G. Nicholson reports consulting fees received from Galapagos, Boehringer Ingelheim, Roche and Medical Quantitative Image Analysis in relation to idiopathic pulmonary fibrosis, outside the submitted work; and payment for educational activities in relation to ILD outside the submitted work.
Conflict of interest: E.A. Renzoni reports receiving grants or contracts from Boehringer Ingelheim outside the submitted work; lecture fees paid to the institution from Boehringer Ingelheim, Roche and Chiesi outside the submitted work; and support for attending ATS conference from Boehringer Ingelheim outside the submitted work.
- Received July 24, 2021.
- Accepted September 1, 2021.
- Copyright ©The authors 2021
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References











Jump To
- Article
- Abstract
- Abstract
- Introduction
- Why was IPAF proposed?
- Advantages of the IPAF concept and definition
- Outstanding issues in the IPAF concept
- Prognostic implications of the IPAF entity
- Should a UIP pattern be excluded from the IPAF entity?
- How does the presence of autoimmune features influence initial evaluation?
- Conclusion
- Acknowledgements
- Footnotes
- References
- Figures & Data
- Info & Metrics