





Abstract
Respiratory diseases are among the leading causes of morbidity and mortality worldwide, representing a major unmet medical need. New chemical entities rarely make it into the clinic to treat respiratory diseases, which is partially due to a lack of adequate predictive disease models and the limited availability of human lung tissues to model respiratory disease. Human pluripotent stem cells (hPSCs) may help fill this gap by serving as a scalable human in vitro model. In addition, human in vitro models of rare genetic mutations can be generated using hPSCs. hPSC-derived epithelial cells and organoids have already shown great potential for the understanding of disease mechanisms, for finding new potential targets by using high-throughput screening platforms, and for personalised treatments. These potentials can also be applied to other hPSC-derived lung cell types in the future. In this review, we will discuss how hPSCs have brought, and may continue to bring, major changes to the field of respiratory diseases by understanding the molecular mechanisms of the pathology and by finding efficient therapeutics.
Abstract
Human pluripotent stem cells may help to develop animal-free, fully human in vitro models to advance our understanding of disease mechanisms, for finding new potential targets by using high-throughput screening platforms, and for personalised treatments. https://bit.ly/3cahaqz
Introduction
Many drugs selected in early drug developmental phases to combat or prevent respiratory diseases fail to meet the strict requirements of clinical studies. This is largely due to a lack of proper models to predict drug efficacy and toxicity in pre-clinical studies [1]. Current pre-clinical models for respiratory medicine include: 1) in vitro models using cell lines or primary cells; 2) ex vivo models using precision cut lung slices; 3) in vivo models using mainly rodents; and 4) in vitro three-dimensional (3D) models such as organoids and lung-on-a-chip models [2, 3]. However, current models do not fully recapitulate human physiology, and the translatability of animal models to the human situation is usually poor. Primary patient material more adequately recapitulates the human situation, but accessibility to primary patient material is often limited, or cells can be difficult to isolate or culture [1, 4]. A recent tool to complement our understanding of respiratory diseases is via the use of human pluripotent stem cells (hPSCs). hPSCs are a combined term referring to human embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). An ESC is a type of pluripotent stem cell isolated from blastocysts, whereas an iPSC is a type of pluripotent stem cell that can be generated directly from a somatic cell. For this, somatic cells are transduced using a set of reprogramming factors, the so-called Yamanaka factors (figure 1) [5–7]. The Yamanaka factors are a set of transcription factors including Oct3/4, Sox2, Klf4 and c-Myc that are commonly transduced using lentivirus into somatic cells like skin fibroblasts or urine-derived renal epithelial cells [8, 9]. In theory, the advantages of hPSCs include their essentially limitless supply, their general accessibility, the possibility to study the impact of rare mutations for personalised medicine, and the possibility to differentiate them to any desired cell type, allowing co-cultures of different cell types from the same patient-derived hPSC line. The past decade has shown that hPSCs are paving the way toward animal-free modelling of diseases in vitro and will potentially further support the acceleration of drug development by enabling high-throughput drug screening, personalised medicine, and cell therapy [5, 6, 10, 11]. This review will provide an in-depth review of the use of hPSCs for disease modelling, drug discovery, toxicology, and cell therapy for respiratory diseases, focusing on the current status of hPSCs in personalised medicine and highlighting their potential and limitations.
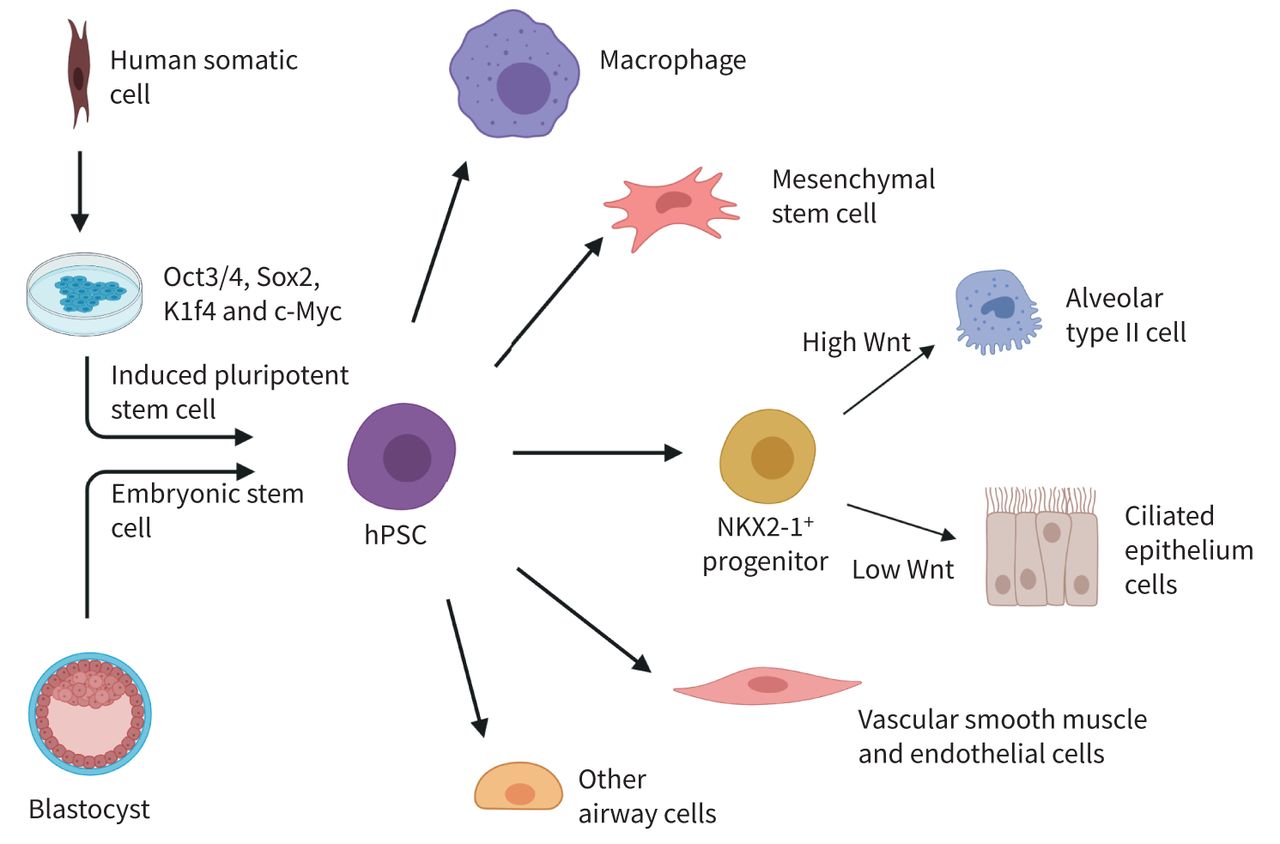
Schematic view of human pluripotent stem cell (hPSC) induction and airway cell differentiation. There are two ways to obtain hPSCs. The first way involves isolating inner cell mass from a blastocyst embryo to obtain embryonic stem cells (ESCs). Another way is by using human somatic cells. These somatic cells are exposed to transcription factors Oct3/4, Sox2, Klf4 and c-Myc to induce induced pluripotent stem cells. ESCs and hPSCs are together referred to as hPSCs. From here, the hPSCs can be differentiated into other cell types among which are a variety of cell types that can be found in the lungs. Figure was generated using Biorender.com.
hPSC differentiation into various lung cell types
The lung is a complex organ containing approximately 58 different cell types [12]. Considering how each cell type contributes to specific functions in a regionally restricted way, the lungs represent a complex organ to model in vitro. Novel hPSC technology may aid in modelling human lung cell types [13]. As depicted in figure 1, the pluripotent ability of these hPSCs can be used to direct cell fate in a specific direction using different concentrations of hormones and growth factors. Cells can be differentiated into distinct lung cell types, together building the complex organ that the lung represents. However, this is a lengthy and complicated process, as each cell type requires the generation of individual protocols. Epithelial cell differentiation protocols exist for both the airways and the alveoli. A small amount of protocols focus on the differentiation of mesenchymal stem cells (MSCs), blood vessel structures, and immune cells, but protocols are still lacking for many airway cells and multicellular airway structures.
Epithelial cells
Epithelial cells are the main cell type studied in hPSC research since they are difficult to capture in vitro from primary cells. This is due to the wide variety of epithelial cell types that play important roles in disease. Hence, epithelial cell differentiation protocols are the most abundant among the available differentiation protocols of lung cells. Several examples can be found in table 1 [14–30]. Identification of Wnt signalling as a key regulator of proximodistal patterning is a critical step for the rapid and reproducible generation from hPSC lines of proximal lung epithelial NKX2-1+ progenitors and their downstream basal, secretory or multi-ciliated progeny, or for the generation of alveolar epithelial cells, as shown in figure 1 [14]. To recapitulate alveolar epithelial development, several stages were mimicked in vitro [15]. Alveolar type II (ATII) cells can be differentiated via NKX2-1+ progenitors under high levels of canonical Wnt signalling [15, 16]. Important markers of mature ATII cells are the presence of surfactant protein B (SP-B) or C (SP-C), and the presence of lamellar bodies. Submerged ATII cultures show the presence of surfactant proteins [15]. It can be argued, however, that immature ATII cells can also express surfactant protein mRNA. The presence of lamellar bodies confirms a fully mature ATII phenotype, which is only observed after the addition of an air–liquid interface (ALI) step; the air exposure step at the luminal side being an additional stimulus for differentiation [16].
Overview of the differentiation protocols for different cell types
Alveolar organoids were generated via NKX2-1+ progenitors as well [17]. Purified hPSC-derived SPC+ cells form monolayered alveolar organoids exhibit self-renewal capacity, and display additional ATII functional capacities, including innate immune responsiveness and the production of lamellar bodies able to package surfactant. It is important to note that the generated alveolar organoids mainly represent fetal ATII cells, and not adult ATII cells, as shown by RNA-sequencing expression profiles. Moreover, the presence of ATII cells was not observed in 3D culture models. ATII differentiation was only observed upon plating of the organoids in ALIs [17]. Next to alveolar organoids, proximal airway organoids were generated from hPSCs, showing ciliary beating in 3D ALI cultures [14, 18]. For the generation of proximal airway organoids, including multi-ciliated airway cells (MCACs) and pulmonary neuroendocrine cells, NKX2-1+ progenitors were treated with fibroblast growth factor-10 (FGF10) and a Wnt activator. ALI culture was crucial for the induced MCACs to demonstrate motile cilia generating flow for mucociliary transport. Alveolar and airway organoids both serve as great tools to study human epithelial cells in vitro, complementary in which air exposure seems crucial to obtain mature cells [14, 18]. The use of ALI cultures optimises in vitro cultures using hPSC-derived epithelial cells, as ALI cultures more closely resemble the air-exchange physiology, mimicking the epithelial function seen in vivo.
MSCs
MSCs are precursors of a variety of structural cells such as airway smooth muscle (ASM) cells and fibroblasts. Protocols for full differentiation of airway structural cells have been lacking so far. Still, protocols for hPSC-derived stromal MSCs have been established. However, the protocols are not abundant and the differentiation methods show high variation, reflecting the high degree of heterogeneity and tissue specificity of MSCs. Different strategies include the addition of FGF-2, platelet-derived growth factor AB, epidermal growth factor or human platelet lysate [19–21], or generating stromal MSCs based on epithelial-to-mesenchymal transition [22]. The generated stromal MSCs are characterised by cell morphology and potency for osteogenic, adipogenic and chondrogenic differentiation [19, 20], and are often referred to as MSC-like cells as they are not tissue specific whereas endogenous MSCs are [21, 22]. Several examples of protocols can be found in table 1. Interestingly, all the above-described protocols did not yield ASM-specific differentiation. Stromal MSCs are precursors of ASM cells as well [31]. The availability of ASM cells would be useful for studies of bronchoconstriction mechanisms and airway hyperresponsiveness.
Endothelial cells and blood vessel structures
Blood vessel structures such as smooth muscle cells (SMCs), endothelial cells (ECs) and pericytes can also be differentiated from hPSCs. Upon obtaining the lateral plate mesoderm, cells were cultured in smooth muscle differentiation medium in the presence of platelet-derived growth factor BB and transforming growth factor-β (TGF-β) for an additional 12 days and mature SMCs were produced [24]. ECs were obtained via the lateral plate mesoderm as well, followed by culture in FGF-2 and vascular endothelial growth factor (VEGF) [25]. In addition, pericytes can be generated to form the microvasculature. Pericytes are also obtained from the mesoderm and directed in the fate of early vascular cells in the presence of VEGF and TGF-β continued with DMEM with 40% fetal bovine serum to induce pericyte maturation [26]. The possibility of differentiating stem cell derived ECs to a lymphatic phenotype characterised by LYVE+ Podoplanin+ expression and positivity for VEGF-A and -C has also been demonstrated, although such cell models have not yet been used extensively for respiratory research [27].
Macrophages and dendritic cells
Macrophages can be differentiated from hPSCs in the presence of colony stimulating factors (CSFs), such as granulocyte-macrophage (GM) CSFs and macrophage (M) CSFs [28, 29]. After co-culturing hPSCs and murine bone marrow stromal cells in the presence of GM-CSF and M-CSF, macrophage precursors were isolated by sorting for CD235a−/CD41a− cells, followed by isolating CD45+ cells. Recovered cells were cultured in the presence of GM-CSF and M-CSF for another 7 days in order to generate mature differentiated macrophages. Mature hPSC-derived macrophages showed functions similar to in vivo macrophages, like tumour necrosis factor-α (TNF-α) release and surfactant clearance [28]. Another method to derive macrophages is via embryoid body (EB) formation, in which EBs were cultivated in the presence of interleukin (IL)-3 and CSFs (GM-CSF, M-CSF or granulocyte (G)-CSF) to generate myeloid cell forming complexes (MCFCs), producing type I macrophages, type II macrophages or granulocytes, respectively. Monocytes, macrophages or granulocytes generated by the MCFCs were harvested from the supernatant. For further maturation, the monocytes, macrophages or granulocytes were cultured for another 7–10 days in the presence of M-CSF, G-CSF or GM-CSF, respectively. Mature macrophages were capable of secretion of TNF-α and several cytokines like IL-8 [29]. The production of macrophages from MCFCs has already been shown to be available for up-scaled production in large stirred-tank bioreactors. MCFCs were induced from EBs in an orbital shaker, in which the MCFCs started to continuously produce immature macrophages that could be harvested weekly for up to 3 months. After another week of maturation, the harvested macrophages represented a homogenous population of CD45+CD11b+ cells with classical macrophage-like morphology and were able to phagocytose Escherichia coli bacteria [32]. Differentiation of hPSCs to macrophages seems to be successful in the presence of M-CSF. However, dendritic cells (DCs; antigen-presenting cells) are still difficult to mimic in vitro. A protocol has been developed to generate DCs but with a low efficiency. For hPSC-derived DCs, hPSCs were cultured in bone morphogenetic protein 4 (BMP-4), stem cell factor, VEGF and GM-CSF. IL-4 was added sequentially in increasing concentrations. By day 20, floating immature DCs were harvested and matured further to CD141+ cells in the presence of GM-CSF, IL-4, interferon-γ (IFN-γ), TNF-α, IL-1β, and prostaglandin E2. However, the levels of the specific lineage markers for CD141+ cells were quite low and the DCs do not surpass the fetal phenotype [30]. An overview of available differentiation protocols is provided in table 1.
In vitro disease modelling and drug screening
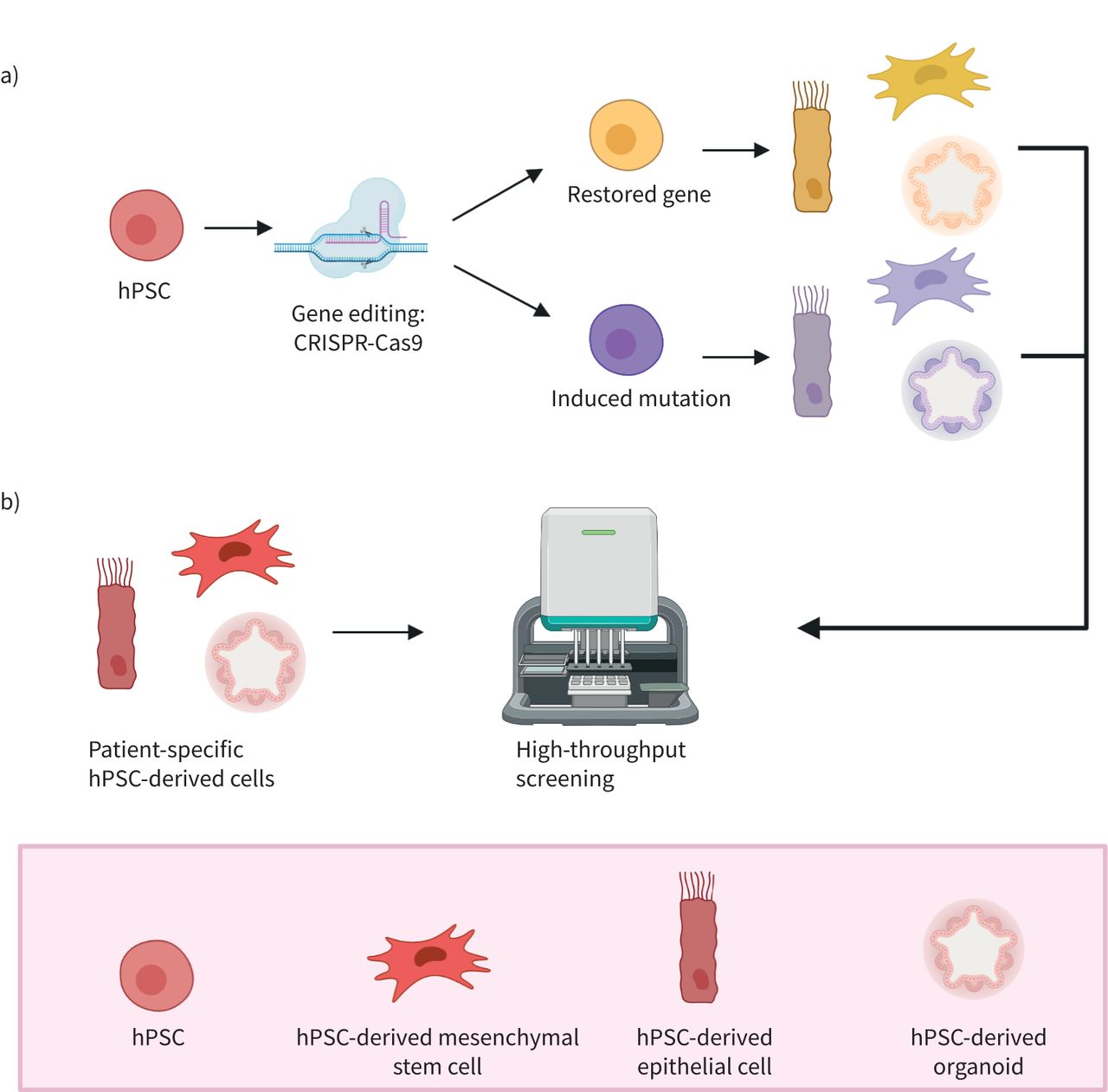
Using hPSC-derived cells, in vitro human disease modelling is now possible for genetic respiratory diseases like cystic fibrosis (CF), and for modelling of pathologies like fibrosis and viral infections, including coronavirus disease 2019 (COVID-19) (see table 2 for examples). In addition, lung organoids have proven to be highly suitable for modelling human lung development in vitro. Figure 2 shows some of the main advantages that hPSCs have over primary cells and cell lines. hPSCs can be genome-edited, both to rescue the diseased phenotype or to incorporate mutations, which helps in the identification of key regulatory genes. Modifying specific genes using, for example, CRISPR-Cas9 to study the mechanisms of disease-dependent genotype is possible in hPSCs without the ethical concerns one would face when working in vivo (figure 2a) [53]. Furthermore, hPSCs aid in drug discovery, for example by implementing high-throughput screening platforms (figure 2b). High-throughput screening platforms using automated equipment can help to rapidly test a high number of samples, facilitating screening for new biomarkers and/or optimal treatment in in vitro disease models [34, 40].
Overview of hPSC differentiation protocols for different diseases and conditions

In vitro modelling using human pluripotent stem cells (hPSCs). Using the advantage of an unlimited supply of hPSCs, several in vitro techniques aid our understanding of disease mechanisms and help accelerate drug development. a) With gene editing techniques, such as CRISPR-Cas9, hPSCs can be edited for inducing a specific mutation, or by restoring a mutated gene. With this, the edited hPSCs can be altered to obtain a certain desired phenotype. b) To accelerate drug testing and development, patient-specific hPSC-derived cells can be used for high-throughput drug screening. In this way, complete libraries of compounds can be tested, optimising the search for a potential drug that could work in a therapeutic application. Figure was generated using Biorender.com.
CF
CF is a respiratory disease that can be caused by mutations in the CF transmembrane conductance regulator (CFTR) gene [54]. Due to its well-understood single-gene mutations, CF was one of the first respiratory diseases studied using hPSCs [55]. Patient-derived hPSCs enable: 1) the study of mechanisms underlying CF in human in vitro models instead of mice; 2) the use of gene technology to repair specific CF mutations; and 3) the use of patient-derived CF-cells and organoids in screens for personalised treatment [55]. Wong et al. [33] were the first to show the potency of in vitro disease modelling using hPSCs as a tool for personalised disease modelling, using hPSC-derived epithelial cells in ALI cultures. Epithelial cells harbouring the F508del mutation (which accounts for 70% of mutations in CF patients) appeared to have a dysfunctional CFTR. When treated in vitro with a small molecule, a so-called corrector compound similar to VX-809, hPSC-derived CF-epithelial cells showed partial restoration of CFTR function. VX-809 is already known to correct CFTR function in patients who are homozygous for F508del [56]. Small patches of CFTR appeared on the cell surface following treatment with the VX-809-like compound C18, while these CFTR channels were absent in untreated cultures [33]. Next, gene correction of the F508del mutation resulted in full restoration of CFTR function in organoids. Compound screening with already known drugs partially rescued CFTR function by treatment with VX-770 and VX-809, as it has been previously shown in CF patients, confirming the usefulness of hPSCs as an accurate human in vitro model [34]. To take the automated compound screening to a next level, halide-sensitive yellow fluorescent protein was included into F508del CF organoids for automated quantitative measurement of CFTR function. Fluorescent quantitative measurement enables high-throughput drug screening platforms that may lead to the identification of novel drugs in a way that is superior compared to screening in cell lines [34]. The study of the monogenic disease CF in hPSCs is more advanced compared to more complex diseases. Current CF research using hPSCs has mainly focused on the predominant F508del mutation, but there is no reason to expect that hPSC-derived CF epithelial cells could not be used for other mutations. Once hPSC models for in vitro disease modelling and drug research have been established for genetic diseases like CF, researchers can look further into using these hPSC-derived models to expand their understanding to more heterogeneous diseases like COPD and asthma.
Rare mutations
Several rare genetic mutations affect epithelial cells and are lethal because of altered surfactant production. SP-B deficiency is a fatal disease affecting neonates, as mutations in the SP-B gene result in a decrease in secretion and an abnormal composition of this surfactant. To demonstrate the effects of genome editing, hPSCs derived from SP-B-deficient patients were altered with a lentivirus carrying the healthy SP-B gene. Both SP-B-deficient and SP-B-corrected hPSCs were differentiated into lung organoids. Corrected organoids showed SP-B protein, the presence of normal lamellar bodies, and secretion of surfactant in the culture medium, suggesting that the mutation can be targeted and modified to rescue the disease phenotype in vitro [35].
Hermansky–Pudlak syndrome (HPS) is a single-mutation disorder that can cause early onset pulmonary fibrosis (PF). Using CRISPR-Cas9, several HPS mutations can be incorporated into hPSCs and subsequently differentiated into lung organoids to induce fibrotic changes. Fibrotic changes were characterised by increased gene expression of collagens and markers for mesenchymal cells, and by extracellular matrix (ECM) deposition. Using these organoids, RNA expression profiles from genome-wide association studies were used to find common upregulated genes that cause fibrosis, like IL-11. IL-11 induced ECM changes in control organoids as well, and its deletion prevented a fibrotic phenotype in HPS-mutant organoids. This way, hPSCs aid in understanding the molecular mechanism of HPS in vitro [36]. Furthermore, hPSCs from HPS patients and their gene-corrected variant were generated and differentiated into ATII cells. Comparing the healthy and diseased populations of ATII cells revealed a difference in the size, dynamics and surfactant secreted from the lamellar bodies of the ATII cells, which indicates that HPS can be studied in ATII cells without the need for patient material [37]. It must be noted, however, that no reports have verified the secretion of pulmonary surfactant in organoids derived from hPSCs or HPS patient-derived cells, making it difficult to prove if the potential drugs could elicit beneficial effects [37].
Some known rare mutations in alveolar macrophages are pulmonary alveolar proteinosis (PAP) syndrome and Mendelian susceptibility to mycobacterial disease (MSMD). PAP is associated with impaired clearance of surfactant by alveolar macrophages [28]. hPSC-derived macrophages from PAP patients and healthy donors both showed a typical morphology and phenotypic markers. However, PAP macrophages showed impaired GM-CSF receptor signalling and reduced surfactant clearance compared to normal macrophages, as was observed in vivo in PAP patients as well. Correcting the GM-CSF receptor signalling improved the surfactant clearance abnormality in PAP macrophages in vitro [28]. MSMD can be caused by genetic mutations of IFN-γ immunity, and MSMD macrophages fail to inhibit mycobacterial growth. hPSC-derived macrophages from patients carrying mutations in their IFN-γ immunity were compared for functionality and inhibition of mycobacterial growth. Indeed, mutated macrophages did not reduce mycobacterial growth when challenged with bacteria, despite their normal phenotype [38]. As demonstrated in the examples above, hPSCs can be used to model the pathogenic molecular and cellular abnormalities in lung cells that drive genetic lung disease.
Lung development and neonatal diseases
Many hPSC-derived epithelial organoids do not fully mature and represent a fetal state [57]. The lung organoids resemble the bronchi/bronchioles of the developing human airway in terms of cells and structure and are surrounded by lung mesenchyme and cells expressing alveolar cell markers. The bud tip progenitor organoids possess a population of highly proliferative multipotent cells with in vitro multilineage differentiation potential. After benchmarking the organoids models, their characterisation showed to be representative of human fetal-like tissue. This is a major problem, as many lung diseases are diseases that are typical for a post-natal or adult lung. It is crucial that the lung airway organoids also represent this post-natal/adult stage to accurately model the disease. There is a need to improve cell maturation. Strategies can include time, medium supplementation, or interaction with other cells to promote cell maturation. For example, for alveolar cells, air exposure might be the way to go to induce maturation of ATI and ATII epithelial cells [16]. Nevertheless, organoids serve as an excellent platform for the study of neonatal and infant diseases, as primary material is mainly available from adult donors. This enables for the first time the study of lung development in vitro in human models, looking at both healthy lung development and defects upon genetic mutations that can affect lung development [57]. Also, the exact nature of the branching process is not yet known. Currently, the process of branching seems spontaneous and space-filling. These organoids can be used to study the mechanism of branching in the future [58].
Pulmonary fibrosis
Fibrosis can be mimicked in mesenchymal organoids using several techniques, without known genetic alterations [39, 40]. A first method to mimic PF in the small airways is by using TGF-β in concentrations that would induce cell alterations. hPSC-derived mesenchymal organoids were cultured in the presence of fibroblast-coated alginate beads, which represented the lung alveolar structure. Organoids treated with TGF-β showed an increased expression of collagen 1 and α-smooth muscle actin (SMA), indicators of fibrotic processes in organoids. Furthermore, the morphological and mechanical effects of PF were mimicked in vitro by an altered shape and increased contraction of the organoid [39]. Secondly, to induce fibrosis by altering the ECM scaffold to culture the cells, MSC-like cells were cultured on stiff (13 kPa) matrices that approximate the stiffness of a fibrotic organ. It was observed that MSC-like cells grown on these substrates showed a highly proliferative phenotype and formed a dense, scar-like structure. Consistently, both the gene- and protein-expression levels of collagen I, α-SMA and TGF-β were upregulated in hPSC-MSC-like cells compared to primary fibroblasts grown on stiff substrates. These hPSC-induced models of fibrosis using hPSC-derived MSCs can help to understand disease mechanisms and aid the identification of novel drugs using high-throughput screening platforms [34].
Viral infection
The human respiratory system is often targeted by viral infections. hPSC-derived organoids and immune cells provide a great toolbox to understand the mechanisms of viral infection. In hPSC-derived DCs, the crucial pathways in innate immune cells upon viral infections could be identified using targeted genome editing. Interferon regulatory factor 5 (IRF5) is a gene suspected to play an important role in the antiviral response. CRISPR-Cas9 was used to generate IRF5-deficient hPSC-derived DCs, which were infected with influenza A. IRF5-deficient DCs showed reduced virus-induced inflammatory cytokine production out impacting virus replication and a reduced type 1 IFN secretion, compared to DCs without the IRF5-mutation. Performing this study in parallel to studies of human lung cells and IRF5-deficient mice demonstrated that IRF5 indeed mediated viral pathogenesis in vivo [41]. Such studies aid the identification of key regulatory genes upon a viral response. Moreover, the infant lung is prone to many viral infections, of which the exact mechanisms are not yet known. Current models to study viral infections consist of primary epithelial cells from the bronchus, obtained from adult donors, so little is known about paediatric response to viruses. Organoids that represent the infant or developing distal lung can be used to recapitulate these disease conditions. Infection of lung organoids with several viral strains led to different responses of the organoids, which were in accordance with the in vivo viral response. For example, infection with parainfluenza virus led to viral shedding without morphological changes, while respiratory syncytial virus infection induced detachment and shedding of infected cells into the lung organoid lumens. Furthermore, infection with childhood viral strains like measles led to syncytium formation in the organoids, showing lung organoids as a representative in vitro model to study infant respiratory diseases [58, 59]. It must be noted, however, that viral infections are simulated by adding viral titres to submerged organoid cultures. Another limitation of working with organoids is that due to the 3D nature of many organoids, the viral particles are offered at the basolateral side of the epithelium. A model with lung organoids derived from adult stem cells showed that the model could be improved when organoids are ALI-cultured, in contrast to the 3D morphology of organoids. This ALI set-up ensures exclusive apical exposure to the virus, similar to the human route of viral infection in the lungs [60]. Interestingly, the aforementioned detachment and shedding of infected cells seen in organoids following respiratory syncytial virus infection was not seen in ALI-cultured epithelial cells, indicating additional differences between these model systems [61].
The current COVID-19 pandemic is a perfect opportunity for the scientific community in hPSC research to show how these hPSC-derived models can aid our understanding of disease mechanisms and drug discovery. For example, hPSC-derived ATII cells on ALI cultures were used for testing antiviral drugs like remesdevir. Simultaneously, the hPSC-derived ALI cultures were compared to in vivo COVID-19 lung autopsies, showing TMPRSS2 protease inhibition in both models. This is a clear example of understanding the importance of both using ALI cultures to mature the ATII cells and simultaneously validating hPSC models with patient material [42]. Next, a high-throughput system was tested in which US Food and Drug Administration-approved compounds were screened on hPSC-derived alveolar organoids. This represents an efficient method to select potential drug candidates for further clinical trials [43]. The development of hPSC models will further aid drug development and facilitate the deeper understanding of COVID-19 pathogenesis.
Pulmonary hypertension
Pulmonary arterial hypertension (PAH) can be modelled using hPSCs by focusing on EC function in the pulmonary arteries. hPSC-derived ECs were validated and compared with primary cells obtained from the same donor. hPSCs showed similar responses in migration, adhesion, and tube formation compared to primary ECs from the same donor. In addition, transcriptomic profiling revealed that high kisspeptin 1 can be related to reduced migration and low carboxylesterase 1 to impaired survival. Given the fact that the cellular responses of primary ECs and hPSC-derived ECs overlap within the same donor, it is proposed that hPSCs are a useful tool in uncovering disease mechanisms and in designing tailored therapies for patients [44]. hPSC-derived SMCs have been studied in relation to PAH as well and compared to an EC response in both control and two isogenic BMP receptor type 2 ( BMPR2)-mutated hPSC lines. Interestingly, in co-cultures of SMCs and ECs, both cell types exhibit a distinct response to the BMPR2 mutation, with the SMCs being more susceptible to heterozygous BMPR2 mutations than the ECs in response to disease-triggering agents like TNF-α [25].
Validation of models
For both hPSC-derived cells and organoids models, human in vitro models will have to be validated before the new models can be integrated into research and drug developmental studies. Many studies validate their human in vitro models in vivo in mice, ex vivo in human slices, and in vitro with primary cells [40, 42, 62]. Another opportunity for validation is by running organoid studies next to clinical trials that have been scheduled already, to get a more accurate validation if human in vivo responses are similar to human in vitro responses [63]. Several clinical trials are currently running for setting up hPSC-supported in vitro models. The INVECCO study (https://clinicaltrials.gov/ NCT03181204) is currently exploring if COPD can be modelled in vitro using iPSCs. Primary bronchial epithelial cells are compared with hPSC-derived epithelial cells, in which COPD is induced using tobacco. Another study (NCT03750734) follows a similar approach to investigate whether iPSCs can help to unravel molecular pathways and identify biomarkers in several lung diseases including idiopathic bronchiectasis, COPD and CF. iPSC-derived epithelial cells will be compared to primary bronchial epithelial cells. One study (NCT02594917) is collecting iPSCs from patients with PAH in parallel with another study involving patients and healthy controls. The PaCyFIC study (NCT03754088) has collected CF-iPSCs and healthy iPSCs as well. In addition, validating iPSC models next to running clinical trials is accelerating and optimising the validation of hPSC models.
Pluripotent stem cells as cell therapy
The clinical potential of hPSCs in respiratory diseases extends even further as they may also be used directly as cell therapeutics. The first in vitro and in vivo animal studies show promise for hPSC-based therapy, contributing to personalised medicine [47–51, 64]. Despite the first successes in vivo in animal studies, cell therapy using hPSCs is not yet available for the patient. This is mainly due to the fact that hPSCs can cause teratoma formation in mice when transplanted into the lungs [65].
hPSC-derived MSCs as a treatment
Co-transplantations of MSCs improve the success rates of lung transplantations. The vulnerability of the transplantation procedure is the rejection of the donor lung caused by an immune response of the recipient. Primary MSCs are known to attenuate immune responses, making them promising tools for use during lung transplantation. Rejection can be prevented, as transplantation of MSCs does not trigger an unwanted immune response (figure 3b) [66, 67]. However, the supply of primary MSCs is limited and cell expansion options are minimal. Differentiation of MSCs from hPSCs for transplantation would ensure the supply of sufficient MSCs. hPSC-derived MSCs can be homogeneously proliferated on a large scale for facilitating the rapid scale-up of clinical applications [68]. Co-transplantation of hPSC-derived MSCs and tracheal tissue in mice showed an attenuated immune response compared to control mice which had a tracheal transplantation without the presence of hPSC-derived MSCs. Furthermore, co-transplantation of hPSC-derived MSCs showed improved oxygenation and blood flow in animal models. These findings demonstrate that administration of hPSC-derived MSCs can act as an immunosuppressor and to improve transplantation success [47].

{kind=link}
{kind=link}
{kind=link}
Transplantation of human pluripotent stem cells (hPSCs) as cell therapy. Transplantation of hPSCs and hPSC-derived cells could be applied in a personalised way in the future. a) hPSCs can be gene-edited using CRISPR-Cas9 to restore a mutated gene. When edited, these hPSCs can be differentiated into the desired cell type and functional cells could be transplanted back into the patient. b) As mesenchymal stem cells are known to attenuate the immune response, hPSC-derived human somatic cells could be used as therapy. These could be transplanted along with a lung transplantation to prevent organ rejection. An alternative use is to help alleviate allergic responses in asthma. c) Transplantation of hPSC-derived epithelial cells and hPSC-derived epithelial organoids into diseased or damaged lung improves and restores the airway function. Figure was generated using Biorender.com.
As hPSC-derived MSCs are immunomodulatory, other inflammatory respiratory diseases benefit from MSC transplantation as well, as illustrated in figure 3b. MSCs have limited efficacy in patients with COPD, but a relatively unexplored area is whether transgenic hPSCs overexpressing certain paracrine factors may help to improve efficacy [69]. Allergic airway inflammation can be reduced by administering hPSC-derived MSCs intravenously in mouse models. Short-term effects of an allergic mouse asthma model included an inhibition of inflammatory cell infiltration and mucus production in the lung and a reduction in eosinophil infiltration in the nose. In addition, in the bronchoalveolar and nasal lavage fluids, there was a decrease in inflammatory cell infiltration plus reduced serum levels of T-helper cell (Th) type 2 immunoglobulins and cytokines (including IL-4, IL-5 and IL-13) [48]. Long-term protective effects of hPSC-MSCs in a chronic mouse asthma model included protection of the mice from the characteristics that are typical for chronic allergic airway inflammation. hPSC-derived MSCs improved airway remodelling and prevented fibrosis. The TGF-β/Smad pathway is crucial in the immunomodulatory effects exerted by hPSC-derived MSCs in a chronic mice asthma model [64]. A PCR array performed in mice treated with hPSC-MSCs was used to identify the key regulatory genes involved in the immunomodulatory mechanisms of these MSCs. In mouse asthma models, chemokine (C-C motif) ligand (Ccl) 11, Ccl24, IL-13, IL-33 and eosinophil-associated, ribonuclease A family member 11 (Ear11) were found to play a prominent role in MSC-immunomodulation [70]. Next to allergic asthma, in steroid-resistant neutrophilic asthma, MSC transplantation was presented as a promising therapy. Systemic administration of hPSC-derived MSCs in a mouse model decreased the levels of Th17 cells and IL-17A. Furthermore, neutrophilic airway inflammation was significantly lower 48 h after ovalbumin and lipopolysaccharide challenge following MSC administration, when compared to control and dexamethasone treatment [49]. All the above studies have been performed in mice and do not show long-term side-effects at least for a period of 2 months. However, these studies did not try to further investigate any potential side-effects 2 months after the systemic administration of the hPSC-derived MSCs. Also, the translation to the human situation is lacking. Clinical trials have already demonstrated the safe use of bone marrow-derived MSCs in asthma patients and hPSC-derived MSCs might be a following step [71].
Epithelial transplantation for regeneration
Epithelial cell transplantation can also serve as personalised transplantation upon acute lung injury for both the upper and the lower airways. hPSC-derived MCACs (hPSC-MCACs), as found in the upper airways, were transplanted into the trachea of rats with surgical defects [50]. Most of the transplanted epithelial cells survived following 1–2 weeks of transplantation, as assessed by counting the cilia beat frequency. This high survival rate of functional epithelial cells makes hPSC-MCACs a promising candidate for tracheal reconstruction [50]. Long-term safety and efficacy studies need to be performed, however, as so far only short-term effects (2 weeks) have been modelled [50]. For established ciliated cells, it has been demonstrated that the half-life is ∼6 months in mouse trachea, after which basal cells are necessary to generate new epithelial cells [72]. As an alternative strategy, hPSCs can be induced towards a basal cell phenotype for transplantation, possibly overcoming this limitation [13].
hPSC-derived ATII administration to mice models with bleomycin-induced lung injury showed that the hPSC-ATII cells differentiated and expressed markers for alveolar epithelial type I cells, which in part can differentiate into ATII cells in situ. Moreover, transplantation of hPSC-derived ATII cells in acute lung injury promoted an increased arterial blood flow into the airways in mice, when compared with control mice without transplantation [51]. In addition, airway delivery of both hPSC-derived ATII cells and lung progenitor cells showed improved lung structure and function after hypoxia-induced bronchopulmonary dysplasia in neonatal mice. The epithelial transplantation resulted in a long-term engraftment without evidence of tumour formation [52]. These studies indicate that hPSC therapy can be safe, as long as cells are no longer in an undifferentiated state. In a mouse model of COPD, transplantation of epithelial cells was more successful, as transplantation of epithelial cells reduced fibrosis and reduced alveolar structural damage [11]. However, transplantation of hPSCs and hPSC-derived epithelial cells has been investigated with little success in conditions of premature birth in mice. hPSCs give rise to teratoma formation, and hPSC-derived epithelial cells did not show any improvement in lung development [73].
For in vivo therapy, hPSCs are an exciting and fairly new way to treat respiratory diseases. Most studies have used them in mice models, and only a few have gone into clinical trials. None of these studies have yet been related to any lung diseases [74]. A promising approach for hPSCs in transplantation is by using differentiated hPSCs, like hPSC-derived MSCs or epithelial cells. However, no human in vivo data exists so far, neither to replicate the transplantation successes in mice, nor to confirm the absence of teratoma formation [64, 75]. Moreover, there are still unknown factors for the transplantation of hPSC-derived cells. For example, the dose of cells for transplantation is not yet known, or whether the engrafted cells are sufficiently representative of the local cells [76, 77].
What is concerning, even for differentiated hPSCs, is that a small fraction of ESCs and hPSCs accumulate dominant-negative mutations in tumour-suppressor genes over time, presenting a cancer risk to patients if hPSC-derived cells are used as a therapeutic. Thus, careful genetic characterisation of hPSCs and their differentiated derivatives should be performed before clinical use [78].
Limitations of hPSCs
In terms of disease modelling, as protocols emerge, is it must be noted that the differentiation of cells and growth of organoids from hPSCs are still expensive, labour-intensive, and face difficulties in obtaining reproducible levels of mature cells with sometimes high degrees of variability between hPSC lines [57, 59, 79]. Even for well-established differentiation of epithelial cells, mature cells were obtained with varying efficiencies [5, 15, 33]. Moreover, the organ- and tissue-specific features of cell types are not always recapitulated using hPSC differentiation. By co-culturing ECs with human intestinal organoids, the ECs obtained a phenotype that appeared more related to native intestinal ECs compared to kidney and lung [80]. Similar advantages are to be expected when co-culturing different lung cells.
Some in vitro models require multiple cell types to be integrated for an improved resemblance to the human in vivo situation. Co-culturing multiple cell types might elucidate disease mechanisms that would not be exposed by culturing either cell type alone. For example, the study of alveolar repair in hPSC-derived epithelial cells without the presence of endothelial or other supporting cells makes it less relevant [35]. Currently, the most accessible way of having multiple cell types in one model is by the generation of lung organoids. For co-cultures of hPSC-derived cells that have been obtained individually, differentiation protocols must be timed in order to be mature together, which can be precarious. For human intestinal organoids, in which the intestinal organoids were cultured together with neural crest cells to develop a nervous system in the organoids, co-culturing both improved maturation of the cell types and revealed cellular interactions between different cell types [81].
As transplantable cells for regenerative medicine, their use could be limited by several factors, including tumorigenic potential, immune rejection, genetic instability, heterogeneity of iPSC lineage phenotypic cells, and lack of full maturity in some iPSC-derived phenotypic cells [82]. Their tumorigenic potential is based on shared iPSCs characteristics with cancer cells, such as unlimited proliferation capacity and the expression of c-Myc, an oncogenic marker. Several strategies currently under investigation focus on reducing or elimination of iPSC tumorigenic potential and include the use of extracellular vesicles released by iPSCs [83], the use of differentiated cells instead of undifferentiated iPSCs, the use of reprogramming transgene-free methods for the generation of iPSCs or c-Myc-independent reprogramming methods by employing small molecules (quercetin and YM155) or pluripotent cell-specific inhibitors (oleic acids) [84].
Conclusion
hPSCs are emerging as a promising model to study respiratory diseases in vitro. They can be differentiated into various lung cell types, aiding in modelling different diseases in vitro. Differentiation protocols for epithelial cells have been established and showed robust ATII differentiation to represent alveoli or recapitulate the beating of cilia in the larger airways. However, protocols for the many other cell types present in the lungs are still scarce. Developing protocols for these non-epithelial cells will be a valuable tool for further drug-developmental studies for the airways, as primary material can be limited in supply. Even though differentiation protocols can be lengthy, hPSCs are applicable in different fields of respiratory disease research. hPSC-derived models can aid in the understanding of disease mechanisms and unravel the pathological genetic signature. High-throughput drug screening platforms can accelerate drug testing and personalised treatments. For the first time, rare genetic mutations can be understood by combining hPSC and gene-editing technologies. The first steps are being taken towards cell therapy using hPSCs. Transplantation of hPSC-derived MSCs and epithelial cells show promising results in mice models. However, beneficial effects are still to be replicated in human in vivo experiments and the safety of hPSC transplantation is still being investigated.
Footnotes
Provenance: Submitted article, peer reviewed.
Conflict of interest: P.A. Goldsteen has nothing to disclose.
Conflict of interest: C. Yoseif has nothing to disclose.
Conflict of interest: A.M. Dolga has nothing to disclose.
Conflict of interest: R. Gosens reports financial support from Aquilo, Boehringer Ingelheim and Longfonds during the conduct of this study and financial support from Sanofi Aventis and Chiesi outside the submitted works.
Support statement: The work was financially supported by the More Knowledge with Fewer Animals (Meer Kennis met Minder Dieren) programme of ZonMW (grant number 114021505) with co-financing from Stichting Proefdiervrij, Aquilo BV, Boehringer Ingelheim and Longfonds. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received February 19, 2021.
- Accepted May 26, 2021.
- Copyright ©The authors 2021
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References










