





Abstract
This review aims to provide an overview of pre-transplant antifibrotic therapy on peri-transplant outcomes and to address the possible role of antifibrotics in lung transplant recipients with chronic lung allograft dysfunction.
Lung transplantation is an established treatment modality for patients with various end-stage lung diseases, of which idiopathic pulmonary fibrosis and other progressive fibrosing interstitial lung diseases are growing indications. Theoretically, widespread use of antifibrotics prior to lung transplantation may increase the risk of bronchial anastomotic complications and impaired wound healing.
Long-term graft and patient survival are still hampered by development of chronic lung allograft dysfunction, on which antifibrotics may have a beneficial impact.
Antifibrotics until the moment of lung transplantation proved to be safe, without increasing peri-transplant complications. Currently, best practice is to continue antifibrotics until time of transplantation. In a large multicentre randomised trial, pirfenidone did not appear to have a beneficial effect on lung function decline in established bronchiolitis obliterans syndrome. The results of antifibrotic therapy in restrictive allograft syndrome are eagerly awaited, but nonrandomised data from small case reports/series are promising.
Abstract
Continuation of antifibrotic treatment during waiting time for lung transplantation proved to be safe. Pirfenidone did not appear to have a beneficial effect in bronchiolitis obliterans syndrome; results of antifibrotic therapy in RAS are eagerly awaited. https://bit.ly/3tgNbnq
Introduction
Interstitial lung diseases (ILDs) encompass a large heterogeneous group of diffuse parenchymal lung diseases, characterised by varying degrees of inflammation and fibrosis. Idiopathic pulmonary fibrosis (IPF) is the most common form of idiopathic interstitial pneumonia and is associated with poor prognosis and a median survival of 2–5 years in untreated patients [1–5]. Despite randomised clinical trials demonstrating use of antifibrotics to slow down disease progression and rate of lung function decline, the usual course of the condition is to progress to end-stage respiratory failure and death [1, 3, 6]. In these patients, and in patients with other end-stage fibrosing ILD, lung transplantation (LTx) can be an established treatment option with the potential to improve survival and quality of life [1, 6, 7].
Pirfenidone and nintedanib: two key antifibrotic drugs
Two antifibrotic compounds, pirfenidone (Esbriet) and nintedanib (Ofev), both proved to be effective in reducing functional decline and disease progression in IPF [8, 9].
Pirfenidone demonstrates combined antifibrotic, anti-inflammatory and antioxidant effects, mediated in part by modulating pro-collagen transcription and suppressing the expression of transforming growth factor-β and consequently reducing fibroblastic cell proliferation and myofibroblast differentiation [3, 10–12]. It is orally active and has a terminal elimination half-life of 2.4 h [13, 14].
Nintedanib is an intracellular tyrosine kinase inhibitor, targeting vascular endothelial growth factor receptor, fibroblast growth factor receptor and platelet-derived growth factor receptor. It inhibits fibroblast proliferation, myofibroblast differentiation and potentially angiogenesis; it has an anti-inflammatory effect [3, 12, 15]. According to the recommendation of the European Medicines Agency, nintedanib should be discontinued before major surgery due to the theoretical possibility of increased perioperative bleeding risk, although there is no guidance on how long before surgery it should be discontinued. It is orally available and has a terminal elimination half-life of 9.5 h [13, 16].
Antifibrotics in IPF
In 2000, IPF was defined as a specific form of chronic, progressive, fibrosing interstitial pneumonia of unknown cause, associated with a histopathological and radiological pattern of usual interstitial pneumonia [2]. The exact pathomechanism is unclear, but chronic subclinical injury seems to induce alveolar damage with impaired repair processes, leading to exuberant fibrosis [3, 17–22] (figure 1a and b). Large randomised clinical trials (CAPACITY-1, CAPACITY-2 and ASCEND for pirfenidone; TOMORROW, INPULSIS-1 and INPULSIS-2 for nintedanib) demonstrated pirfenidone and nintedanib to be effective in delaying disease progression, progression-free survival, and for nintedanib, time to first acute exacerbation in IPF [3, 20]. A risk reduction for IPF-related mortality was seen with pirfenidone, and a similar trend was observed with nintedanib [23–27]. The European Union approved the use of pirfenidone for mild to moderate IPF in 2011, and nintedanib for all stages of IPF in 2015. In the United States, the Food and Drug Administration (US FDA) approved pirfenidone and nintedanib for the treatment of IPF in 2014.

{kind=link}
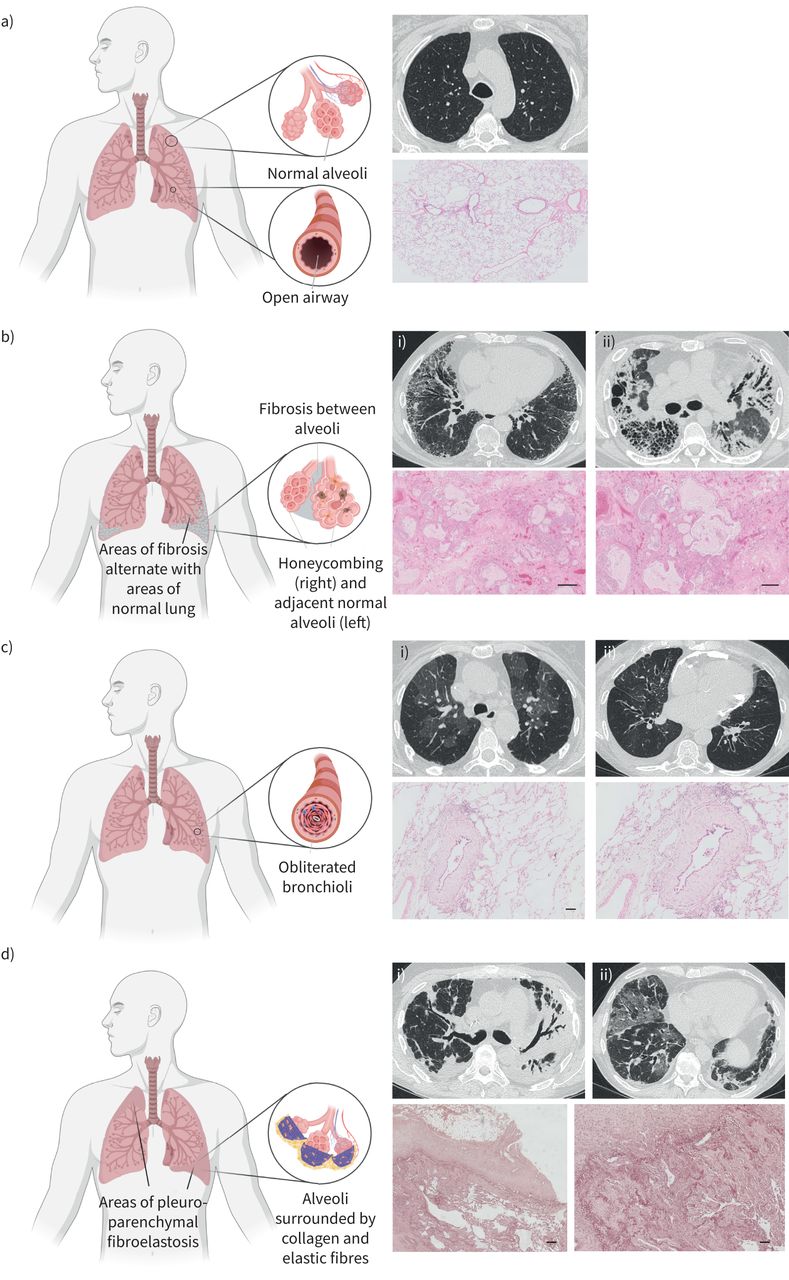
Overview of normal lungs, idiopathic pulmonary fibrosis (IPF), bronchiolitis obliterans syndrome (BOS) and restrictive allograft syndrome (RAS). a) Normal lungs: chest computed tomography (CT), axial pulmonary window: normal lung parenchyma. Haematoxylin and eosin (HE) staining illustrates normally preserved lung parenchyma. b) IPF: chest CT, axial pulmonary window: i) peripheral subpleural reticulation and traction bronchiectasis; ii) honeycombing (right lower lobe), diffuse areas of consolidation and bronchiectasis. HE staining illustrates prominent honeycombing, characterised by irregular cystic airspaces of varying size, partly filled with mucus and surrounded by fibrotic remodelling. c) BOS: chest CT, axial pulmonary window: i) air trapping; ii) bronchiectasis. HE staining with presence of a typical bronchiolitis obliterans lesion, characterised by concentric subepithelial fibrosis narrowing the airway lumen. d) RAS: chest CT, axial pulmonary window: i) bilateral opacification especially of the left upper lung lobe, volume loss of the left lung, traction bronchiectasis; ii) areas of ground-glass opacification and interlobular septal thickening. Elastica van Gieson stain highlights the presence of prominent pleural fibrosis and subpleural fibroelastosis, consistent with pleuroparenchymal fibroelastosis. Figure partially created with BioRender.com.
Despite these positive findings, IPF remains a progressive and ultimately fatal disease, for which LTx is the only definite treatment option in well-selected patients [1, 3, 6, 7, 21]. Furthermore, side-effects (typically gastrointestinal symptoms for pirfenidone and nintedanib; photosensitivity for pirfenidone) or adverse events (mainly toxic hepatitis) may lead to dose reduction, treatment interruption or discontinuation, which again may accelerate disease progression [20, 28]. Hence, early evaluation and referral of possible candidates for LTx are highly recommended [29].
Over the past two decades, idiopathic interstitial pneumonia, of which IPF is the most prevalent form, has become the most frequent indication for LTx (32.4%) [30]. This is partly due to the implementation of the Lung Allocation Score, prioritising patients most in need of transplantation and most likely to benefit. Dependent on age, comorbidities, local experience and donor availability, both single LTx and bilateral LTx are performed in patients with IPF [3, 31].
Antifibrotics in non-IPF progressive fibrosing ILD
ILD comprises a large group of >200 parenchymal lung diseases. A proportion of patients with non-IPF ILD (including ILD associated with rheumatoid arthritis, systemic sclerosis, polymyositis/dermatomyositis, chronic sarcoidosis, chronic hypersensitivity pneumonitis, idiopathic nonspecific interstitial pneumonia and unclassifiable ILD) may develop a progressive fibrosing phenotype causing progressive respiratory symptoms, lung function decline, limited response to immunomodulatory therapies and, potentially, early death [32–35].
There are promising data for antifibrotic therapy in these patients. In the SENSCIS trial, nintedanib reduced the rate of disease progression in patients with systemic sclerosis-associated ILD [36]. The INBUILD trial demonstrated efficacy of nintedanib in attenuating disease progression in patients with various forms of non-IPF progressive fibrosing ILD [37]. Pirfenidone has been studied in a range of progressive non-IPF ILD in the RELIEF study, which was terminated early due to low recruitment, with the final publication awaited [34]. Furthermore, the unclassifiable ILD study in which the primary end-point was home spirometry could not be analysed, but the classical end-points of pulmonary function evolution over time were positive [38]. Several other studies, including the use of pirfenidone in progressive fibrotic sarcoidosis (PirFS) and in pulmonary fibrosis with anti-myeloperoxidase antibodies (PIRFENIVAS), and nintedanib in progressive pneumoconiosis (NiPPS), are ongoing [34, 38].
Based on these available data, nintedanib has been approved for patients with systemic sclerosis-associated ILD and patients with chronic fibrosing ILD with a progressive phenotype by the US FDA and the European Medicines Agency.
An increasing number of LTx have been reported for these patients. According to the International Society for Heart and Lung Transplantation registry, 8.1% of LTx were performed for this indication in 2017 compared to 4.4% in 2007 [30].
Antifibrotics in ILD patients referred for LTx
Effect on listing
IPF is generally recognised as a LTx indication with high waitlist mortality compared to other diagnoses, probably due to its unpredictable clinical behaviour [39–41]. Some patients decline gradually, while others progress rapidly, and some have periods of relative stability punctuated by acute exacerbations [1]. In candidates enrolled for LTx, rapid deterioration can lead to death while awaiting a suitable donor [3]. Deciding on referral and listing for LTx has become even more complicated as antifibrotics alter disease progression, and physicians may choose to wait longer, especially in patients with moderate disease, to await treatment response [1, 28]. However, the use of antifibrotics decreases the risk of acute exacerbation, but does not nullify this risk. The fact that antifibrotics attenuate disease progression should not imply that LTx is no longer needed, but may only be postponed. An unintended effect of antifibrotic treatment could be to delay the need for transplantation beyond age of eligibility, as many centres consider age >65 years a contraindication for (bilateral) LTx [1, 28]. One wonders whether possible LTx candidates should not receive antifibrotics instead, or whether there is a possibility to transplant well-selected IPF patients without comorbidities at a later age, both of which raise ethical doubts.
It is currently unclear whether antifibrotic treatment, when effectively achieving disease stabilisation, influences the Lung Allocation Score [28]. A small retrospective Japanese study demonstrated a significant reduction in Lung Allocation Score in a group of IPF patients with pirfenidone awaiting LTx versus those without pirfenidone [40]. Conversely, a small study of nine IPF patients treated with antifibrotics by Delanote et al. [28] documented unchanged functional exercise capacity and Lung Allocation Score while on the waitlist.
Effect on peri-transplant outcome
Pirfenidone and nintedanib attenuate fibroblast activity, and administration prior to LTx may theoretically increase the risk of bronchial anastomotic complications and impaired wound healing [1, 13, 39, 40]. Through inhibition of vascular endothelial growth factor, nintedanib could also increase perioperative bleeding risk, and the European Medicines Agency suggests cessation during the perioperative period [1, 16]. However, stopping antifibrotic therapy on the transplant list might accelerate disease progression and thus the risk of death [13, 42].
In clinical practice, no evidence has been reported that pre-transplant antifibrotic therapy up to LTx is associated with increased complications [1]. The largest retrospective multicentre study by Mackintosh et al. [42] showed in general no significant difference in bronchial dehiscence (although there were significantly more bronchial dehiscences in the first 6 weeks in the antifibrotic group), 30-day and 1-year survival in 226 IPF patients of whom 40 received antifibrotics at time of LTx (29 pirfenidone, 11 nintedanib). Another multicentre study by Lambers et al. [43] with 132 patients, of whom 36 received antifibrotics (23 pirfenidone, 13 nintedanib), demonstrated no difference in use of blood products, haemothorax needing surgical revision, wound infection and 1-year outcome. A small multicentre study by Balestro et al. [39] focusing on nintedanib in nine IPF patients noted no increased risk of bleeding, impaired wound healing or bronchial anastomotic dehiscence. Leuschner et al. [13] found no influence of antifibrotics on wound and anastomotic healing, blood product use, surgical revision, acute cellular rejection, chronic lung allograft dysfunction (CLAD) or survival (23 pirfenidone, seven nintedanib, mean follow-up 24 months). Several other smaller single-centre studies with varying outcome points could not demonstrate a correlation between pre-transplant antifibrotics and airway dehiscence, impaired wound healing, or bleeding events [28, 40, 44–47].
Furthermore, Veit et al. [31] demonstrated a possible beneficial effect in IPF patients treated with pirfenidone undergoing single LTx, with shorter length of mechanical ventilation and intensive care stay, less severe primary graft dysfunction at 72 h, and lower incidence of acute cellular rejection within the first 30 days. Zhang et al. [48] also reported that IPF patients treated with pirfenidone had less histopathological evidence of acute lung injury (less diffuse alveolar damage and organising pneumonia) at time of LTx, and a trend towards less lymphocytic interstitial infiltration in lungs of patients treated with nintedanib.
One hypothesis to explain the lack of clinical effects (although the pathophysiological mechanisms influence pathways of wound healing) are the short half-lives of pirfenidone and nintedanib, 2.4 h and 9.5 h, respectively [14, 16, 42]. Furthermore, there are no concerns about drug–drug interactions. Hepatic metabolism of pirfenidone primarily occurs through CYP1A2 enzyme (calcineurin inhibitor trough levels are mainly altered by CYP3A4 metabolism), whereas nintedanib is a substrate of p-glycoprotein and only weakly interferes with CYP3A4 [14, 16].
To conclude, antifibrotics until the moment of LTx proved to be safe, having no significant effect on suture complication, bleeding or re-intervention rates, and with possible beneficial effects on primary graft function. The current accepted practice therefore is to continue antifibrotic therapy up to LTx.
Antifibrotics after single LTx
No consensus exists for use of antifibrotics post-LTx and most centres advocate discontinuation. However, one might suggest that patients with IPF undergoing single LTx might benefit from continuation to attenuate fibrosis progression in the native lung [1]. Conversely, recent studies reported no differences in survival between bilateral LTx and single LTx for patients with IPF, and therefore there might be no added value from antifibrotics [49–51]. The first results of a single-centre randomised controlled trial investigating continuation of nintedanib after single LTx in IPF subjects are expected in December 2021 (NCT03562416).
Mechanisms of antifibrotics on the pathophysiological processes after LTx and in CLAD development
Long-term graft and patient survival after LTx are still hampered by development of CLAD, affecting up to 50% of patients within 5 years post-transplant [30]. Both phenotypes of CLAD, bronchiolitis obliterans syndrome (BOS) and restrictive allograft syndrome (RAS), are associated with fibroproliferative features, which may be caused by increased release of growth factors, such as fibroblast growth factor, platelet-derived growth factor and vascular endothelial growth factor [52–56]. Adequate treatment of CLAD remains a significant unmet need. Based on the mechanisms of action, there is a particular interest in the use of antifibrotics in these patients, as pirfenidone and nintedanib could reduce development of obliterative and constrictive bronchiolitis, the histological correlate of BOS, and parenchymal and interstitial fibrosis, the histological correlate of RAS [57].
Pirfenidone has both antifibrotic and anti-inflammatory effects and may, subsequently, have lung allograft protective properties. These effects are believed to be mediated through inhibition of expression of pro-fibrotic and pro-inflammatory cytokines, and enhanced expression of anti-inflammatory cytokines [31, 58]. Several reports demonstrated graft-protective effects and reduced development of fibrosis in tracheal, lung and cardiac animal transplant models [58–63]. In addition to its antifibrotic effects, animal models demonstrated the ability of pirfenidone to limit alloimmune inflammatory responses by impairing T-cell activation and proliferation, and by inhibiting effects upon dendritic cell activation, maturation and function [31, 58, 63, 64]. Moreover, it exhibits antioxidant properties by scavenging reactive oxygen species and preventing lipid peroxidation [65, 66]. These effects have elicited interest in its use as a potential therapeutic agent to prolong allograft survival by inhibiting both fibroproliferative and alloimmune responses [58].
In contrast to pirfenidone, far fewer in vitro and in vivo data are available on nintedanib. In a rat model of left-lung allotransplantation, no reduction in fibrotic changes was observed in nintedanib-treated allografts compared to non-treated allografts, despite reduced expression of growth factors. Early and sustained treatment did not decrease the number of rats with histological signs of acute or chronic rejection [57].
Antifibrotics in CLAD phenotype BOS
BOS is the most common presentation form of CLAD, diagnosed in ∼70% of CLAD patients [52]. The exact pathophysiological mechanism is not yet fully understood, but is widely perceived to be associated with the production of growth factors and inflammatory cytokines, alloimmune processes, non-alloimmune processes and autoimmune processes [65, 67–69]. The histological hallmark of BOS is obliterative or constrictive bronchiolitis, which is thought to arise as lymphocytic bronchiolitis, followed by fibroproliferative obliteration of small airways, leading to segmental subtotal or total obliteration of the lumen of small airways (figure 1c). In advanced stages, a completely acellular fibrotic obliteration is seen, leaving only a scar of the bronchiolar lumen (so-called vanishing airways disease) [69, 70].
Transbronchial biopsies and autopsies from deceased BOS patients revealed interstitial fibrosis next to obliterative bronchiolitis scars, and might thus suggest a role for antifibrotics in BOS [71]. Pirfenidone reduced graft obliteration in animal transplant models [59–61, 67]. Apart from a case study and small case series with favourable results, few real-life results were known until recently [72, 73]. However, a large, multicentre, randomised, double-blind placebo-controlled European trial of pirfenidone in new-onset progressive BOS patients (EPOS) could not demonstrate a significant beneficial effect of pirfenidone on pulmonary function decline [71]. So far, no data regarding the effects of nintedanib in BOS are available. A multicentre, randomised, double-blind placebo-controlled trial of nintedanib in LTx recipients with BOS grade 1–2 is ongoing (INFINITx-BOS), assessing the efficacy of nintedanib in reducing the rate of decline of forced expiratory volume in 1 s as primary end-point. The primary completion date is estimated to be November 2022.
Therefore, the use of pirfenidone in BOS is currently not recommended, except in clinical trials. Since there are more extensive fibrotic abnormalities, similar to IPF, in patients with RAS, we might perhaps expect a greater effect from antifibrotics in those patients.
Antifibrotics in CLAD phenotype RAS
RAS has been described recently as a novel phenotype of CLAD and occurs in up to 30% of CLAD patients [52]. It is characterised by a restrictive pulmonary function, persistent pleuroparenchymal abnormalities on computed tomography (e.g. ground-glass opacities, consolidation, interstitial reticular thickening, (sub)pleural reticulation, traction bronchiectasis, architectural distortion) and diffuse alveolar damage, focal fibroblastic foci, pleuroparenchymal fibroelastosis and concurrent obliterative bronchiolitis on histology [74, 75] (figure 1d). The pathomechanisms in RAS are thought to be, at least partially, similar to those in IPF. Therefore, antifibrotics may be uniquely suited agents in the management of RAS [75]. Some case reports and one small case series have demonstrated attenuation in the rate of pulmonary function decline with off-label use of pirfenidone or nintedanib [75–77]. Larger randomised trials are pivotal and pending. The open-label trial PIRCLAD, in which pirfenidone has been investigated in LTx patients with restrictive CLAD, showed it in preliminary analysis to be safe and tolerable; results regarding efficacy are expected in October 2021 [78]. The results of STOP-CLAD, a randomised, double-blind placebo-controlled trial investigating the effects of pirfenidone in patients with BOS or RAS are expected any time. The primary end-point is to evaluate the change in percentage of functional small airways disease as measured by parametric response mapping; changes in pulmonary function tests (forced expiratory volume in 1 s, forced vital capacity) are secondary end-points.
Future areas of interest
Antifibrotics in pulmonary chronic graft-versus-host disease
Long-term survival after allogeneic haematopoietic stem cell transplantation (alloHCT) is limited by chronic graft-versus-host disease (cGvHD) [79, 80]. Pulmonary cGvHD, which can present with obstructive and/or restrictive changes, is thought to affect 5% of alloHCT recipients, but this prevalence is probably underestimated due to underdiagnosis [81]. Therapeutic options are limited; several therapies have been adopted from CLAD after LTx, but use of these drugs remain mainly empirical [82–84].
BOS phenotype
BOS is the most well-defined late complication and the only entity formally recognised as manifestation of pulmonary cGvHD. It is characterised by progressive small-airway disease, often failing to respond to treatment, leading to respiratory insufficiency and death [81, 84]. BOS after alloHCT is probably caused by similar pathophysiological mechanisms to those occurring in BOS after LTx, leading to inflammation of small airway epithelial cells and subepithelial structures, excessive fibroproliferation and progressive airflow obstruction [81, 82]. Pirfenidone has shown to be effective in a murine model of cGvHD by reducing macrophage infiltration, transforming growth factor-β production and T-cell reaction [64]. A recent case report noted a remarkable improvement of respiratory symptoms and pulmonary function after starting nintedanib in a patient with BOS post-alloHCT, suggesting that nintedanib may be a possible novel therapeutic option [85]. Hostettler et al. [86] evaluated the effect of pirfenidone in five alloHCT patients with BOS; however, no follow-up results are known. Prospective trials are warranted; the results of an open-label study investigating nintedanib in patients with BOS following alloHCT (NINBOST2018) are expected in 2021. Nevertheless, there is a chance that these results will be negative, corresponding with the results of the EPOS trial investigating pirfenidone in BOS after LTx, and we might perhaps expect more effect from antifibrotics in restrictive forms of cGvHD.
Interstitial phenotype
Recent studies showed other presentation forms of pulmonary cGvHD in 12–60% of patients, such as cryptogenic organising pneumonia and ILD, resulting in restrictive lung function impairment. Overall, the clinical presentation is comparable to RAS after LTx [82–84, 87]. However, clear definitions of these entities are still lacking, making exact diagnosis and targeted treatment difficult.
To our knowledge, there is only one clinical report describing efficacy of nintedanib in fibrotic lung disease after alloHCT. This report described two patients, of whom one had beneficial effect of nintedanib on fibrotic pulmonary cGvHD; the other patient had persistent fibrotic changes after alloHCT due to disease-related diffuse alveolar haemorrhage, but also improved with nintedanib [88]. Large studies are needed to investigate this further.
Antifibrotics in other solid-organ transplants
By extrapolation, the question arises whether antifibrotics may influence fibrotic allograft dysfunction in other solid-organ transplant recipients. However, to date, virtually no research has been conducted in this area. We could only find one study describing renoprotective effects of pirfenidone on chronic renal allograft dysfunction by reducing inflammation and renal fibrosis in a rat model [89]. It is likely that further results on antifibrotics in CLAD will have to be awaited before investigation in this area will be conducted.
Inhaled antifibrotics
Pirfenidone and nintedanib have their limitations as oral drugs. Both drugs were reformulated as a solution for inhaled administration, which holds promise to reduce systemic side-effects, maximise local efficacy and possibly enable dose escalation for additional efficacy in IPF patients [90, 91]. Inhaled pirfenidone and nintedanib proved to have good pharmacokinetics, delivering an oral-equivalent lung maximum concentration with lower systemic levels, and to be well-tolerated and effective in several animal models [90–92]. These promising observations have to be confirmed in human trials. Aerosolised pirfenidone already appeared to be well tolerated in healthy subjects and IPF patients [93].
Once more is known about inhaled antifibrotics in IPF, their use might be adopted in LTx patients with CLAD, with the benefit of fewer side-effects and direct deposition in the lung allograft.
Conclusion
Antifibrotics until the moment of LTx proved to be safe, having no significant effect on bronchial anastomotic healing, wound healing, bleeding or re-intervention rates. The accepted practice is to continue antifibrotics until time of LTx. Currently, few results are known about the use of antifibrotics in CLAD. The EPOS trial appeared to be a negative study in which no effect of pirfenidone on the course of BOS could be demonstrated. However, the results of antifibrotic therapy in patients with RAS, where antifibrotics may be of greater benefit, are eagerly awaited.
Footnotes
Provenance: Submitted article, peer reviewed.
Author contributions: S. Bos performed research, collected data and wrote the manuscript. L.J. De Sadeleer, J. Yserbyt, W.A. Wuyts, H. Beeckmans and A. Sacreas critically revised the manuscript. A. Vanstapel provided the anatomopathology images and critically revised the manuscript. R. Vos coordinated and designed the research, collected data, and critically revised the manuscript.
Ethics approval: Ethics Committee of the University Hospital of Leuven under agreement S51577. Patient provided written informed consent at time of listing for transplantation to access her clinical and biobanked data for research purposes.
Conflict of interest: S. Bos has nothing to disclose.
Conflict of interest: L.J. De Sadeleer reports non-financial support from Roche and Boehringer Ingelheim, outside the submitted work.
Conflict of interest: A. Vanstapel has nothing to disclose.
Conflict of interest: H. Beeckmans has nothing to disclose.
Conflict of interest: A. Sacreas has nothing to disclose.
Conflict of interest: J. Yserbyt has nothing to disclose.
Conflict of interest: W.A. Wuyts reports grants from Boehringer Ingelheim and Roche, paid to his institution, outside the submitted work.
Conflict of interest: R. Vos reports grants from Research Foundation Flanders (FWO), outside the submitted work.
Support statement: The following authors are supported by a research fellowship, but received no specific funding for the current study: R. Vos and W.A. Wuyts are senior clinical research fellows of the Fund for Scientific Research Flanders (FWO); A. Vanstapel is supported by a fundamental research grant from the Fund for Scientific Research Flanders (FWO)(1102020N).
- Received February 25, 2021.
- Accepted April 2, 2021.
- Copyright ©The authors 2021
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References











Jump To
- Article
- Abstract
- Abstract
- Introduction
- Pirfenidone and nintedanib: two key antifibrotic drugs
- Antifibrotics in IPF
- Antifibrotics in non-IPF progressive fibrosing ILD
- Antifibrotics in ILD patients referred for LTx
- Mechanisms of antifibrotics on the pathophysiological processes after LTx and in CLAD development
- Antifibrotics in CLAD phenotype BOS
- Antifibrotics in CLAD phenotype RAS
- Future areas of interest
- Conclusion
- Footnotes
- References
- Figures & Data
- Info & Metrics