





Abstract
Nontuberculous mycobacteria (NTM) are diverse microbial species encompassing commensals and pathogens with the ability to cause pulmonary disease in both immunocompetent and immunocompromised individuals. In contrast to Mycobacterium tuberculosis, which has seen a reduction in disease rates in developed countries, the incidence and prevalence of NTM disease is increasing. NTM are difficult to treat with standard antimicrobial regimens and may contain both virulence and antibiotic-resistance genes with potential for pathogenicity. With the advent of molecular techniques, it has been elucidated that these organisms do not reside in isolation and are rather part of a complex milieu of microorganisms within the host lung microbiome. Over the last decade, studies have highlighted the impact of the microbiome on host immunity, metabolism and cell–cell communication. This recognition of a broader community raises the possibility that the microbiome may disrupt the balance between infection and disease. Additionally, NTM disease progression and antimicrobial therapy may affect the healthy steady state of the host and function of the microbiome, contributing to further dysbiosis and clinical deterioration. There have been limited studies assessing how NTM may influence the relationship between microbiome and host. In this review, we highlight available studies about NTM and the microbiome, postulate on virulence mechanisms by which these microorganisms communicate and discuss implications for treatment.
Abstract
NTM disease is increasing in prevalence. The lung microbiome represents an ecological niche of dynamic interplay unique to each individual and plays a potential role in NTM disease. https://bit.ly/3ln0D67
Introduction
The Mycobacterium genus is a diverse group of both pathogenic and commensal organisms with over 190 distinct species recognised. Certainly, the species with the greatest impact thus far on human health from a clinical, societal and historical perspective is Mycobacterium tuberculosis. However, over the last three decades, emerging epidemiological evidence has highlighted that both the incidence and prevalence of other species from the Mycobacterium genera, referred to as nontuberculous mycobacteria (NTM), are increasing. Given the increased incidence of NTM, clinical awareness of the impact of these organisms in disease states and health has heightened. Furthermore, the human lung microbiome has emerged as a complex ecological community comprising commensals, symbiotic microorganisms and pathogens. The complex interplay between infection and disease with NTM in the human microbiome is currently unclear and the focus of this review.
Nontuberculous mycobacteria
NTM are aerobic non-motile organisms belonging to the Actinobacteria phylum, ubiquitously found in water and soil [1, 2]. While geographic location affects the prevalence of specific NTM, pathogens comprising the Mycobacterium avium complex (MAC) are the most common worldwide, followed by Mycobacterium abscessus and Mycobacterium kansasii [3, 4]. Exposure to NTM is widespread because they are environmental organisms; however, very few exposed individuals develop infection or progress to fulminant lung disease, suggesting that NTM disease can be attributed to various host factors [5]. These factors can be categorised into broad groups, including 1) structural lung abnormalities, 2) immune or genetic disorders that predispose to disease, 3) connective tissue disease features and 4) no clear predisposing conditions, often with a predilection in frail older women; however, these categories are not mutually exclusive [1, 2, 6, 7].
In developed countries, the disease burden of NTM infection has surpassed the prevalence of M. tuberculosis, the pathogen causing tuberculosis (TB) [8, 9]. Reasons for increased prevalence rates are multifactorial and include growing populations of at-risk individuals, enhanced virulence, altered host–pathogen interactions, more common use of medical immunosuppression and heightened clinician awareness [8, 10]. While epidemiology related to NTM is limited because disease reporting is not considered mandatory worldwide, the annual prevalence of pulmonary NTM infection in the USA ranges from 13.9 to 48 cases per 100 000 persons based on geographic location [11, 12], with similarly increasing NTM-related deaths [13]. Over a similar time period, the prevalence of non-cystic fibrosis (CF) bronchiectasis has also increased in the USA, with an estimated prevalence of 139 cases per 100 000 persons and up to 812 cases per 100 000 persons in those ≥75 years of age [14].
Diagnosis of NTM pulmonary disease can be challenging for clinicians. In part, this may be due to the ubiquitous presence of the organisms in the environment, with identification of the organism on clinical sampling not indicative of lung disease but rather reflecting either transient or permanent infection [15–18]. Infection may progress to pulmonary disease in certain individuals owing to mycobacterial and host factors. Diagnosis is based on a combination of clinical, radiographic and microbiological criteria outlined by the American Thoracic Society (ATS) and the Infectious Diseases Society of America (table 1) [19].
American Thoracic Society/Infectious Diseases Society of America criteria for diagnosing NTM pulmonary disease [19]
The respiratory microbiome and the landscape of microbial players
The human microbiome is an ecological niche of complex microbial communities including bacteria, viruses, fungi and archaea. Infections are a major cause of morbidity in non-CF bronchiectasis. Prior culture-dependant techniques primarily focused on selective growth of a restricted range of bacterial species in clinical samples [20], which historically was the basis of clinical management. However, growing evidence of complex polymicrobial communities in pulmonary disease is now appreciated. These communities have the potential to alter virulence properties or modulate antimicrobial susceptibility towards targeted therapy. Complete characterisation of these microbial communities is essential to better understand interactions and ascertain potential clinical consequences [21]. The gold standard of direct airway sampling is considered to be lower respiratory tract bronchoalveolar lavage (BAL). However, BAL is invasive, time-consuming and associated with clinical risks for sample acquisition. Sputum samples, expectorated or induced, are often used as a surrogate of the lower respiratory tract microbial community and may be optimal based on the clinical question and setting. Concerns in the airway microbiome literature have arisen from potential contamination of sputum from oropharyngeal sources such as the mouth, sinuses and pharynx [22]. Studies in CF have demonstrated contamination of sputum samples from the oropharynx to be nonsignificant, with specific organisms, such as anaerobes, cultured from sputum samples found primarily in the lungs and not the oral cavity [23, 24]. Culture-independent techniques that describe the composition of bacterial communities, including high-throughput sequencing using the 16S ribosomal RNA (rRNA) gene, have substantially advanced microbiome research over the last decade with the ability to detect more microbes from the same clinical specimens than standard culture techniques [20, 25]. Despite advances in microbial communities with pyrosequencing, this technique is not yet available for routine clinical use, primarily owing to the cost and time-consuming nature of analysis.
While composition of the microbiota has been identified in many disease states, the link between commensal changes and specific disease progression has not been well established. Previously thought to be a sterile environment, lungs normally harbour a diverse microbiome of low microbial biomass that is composed of Prevotella, Streptococcus, Veillonella, Fusobacterium and Haemophilus genera [26, 27]. The composition of the lung microbiome is determined by a balance between microbial migration from the upper respiratory tract and oral cavity and microbial elimination by the host by mucociliary transport, cough and antimicrobial properties of the innate and adaptive immune system [26, 28]. In diseased states this balance is often altered, typically leading to an increased microbial load and decreased diversity of species [26].
In patients with non-CF bronchiectasis, the disease cycle is characterised by airway obstruction, host inflammation with bacterial colonisation and sequelae of disease that propagate periods of acute clinical exacerbation and pulmonary function decline [25, 29]. Traditional culture-based techniques using classical aerobic-based techniques do confirm the presence of typical dominant pathogens such as Haemophilus influenzae, Pseudomonas aeruginosa and Streptococcus pneumoniae [30], coexisting with an incredible diversity of satellite taxa in the lower airways of non-CF bronchiectasis patients. Notably, however, in many patients these traditional techniques do not result in the microbiological identification of organisms, despite repeat sampling. Culture-independent methods of sputum sampling demonstrate a complex and diverse microbial community including both aerobic (e.g. Achromobacter, Stenotrophomonas and Streptococcus) and anaerobic (e.g. Prevotella, Veillonella and Actinomyces) genera during periods of clinical stability [22]. While culture-independent techniques confirm the presence of a “core” microbiome composed of similar organisms to those found by culture-dependent techniques (H. influenzae, P. aeruginosa and S. pneumoniae), more than 140 species have been identified, with many species unlikely to be reported through standard diagnostics [31]. Sequencing of bronchiectasis patient sputum samples reveals complex communities that are on a continuum but that can be broadly partitioned into two distinct groups, one with a relatively small number of organisms dominating the community (most often Haemophilus, Pseudomonas and Streptococcus) and a second with less “conventional taxa” with higher diversity and lower abundance [22]. These findings of patient-specific community structures dominated by a single or few genera, regardless of the classification, is consistent with other respiratory diseases that highlight distinctive individual profiles over time [22, 32–34]. These profiles, while unique on an individual level, do fluctuate during times of clinical stability and acute exacerbation.
While causative relationships between the dynamics of the microbial community and clinical outcomes are unclear, several correlations have been observed. A study of 70 non-CF bronchiectasis patients initially assessed standard microbial culture from sputa [35]. 51 patients (73%) were culture-positive for pathogenic microorganisms, most commonly P. aeruginosa (33%) and H. influenzae (21%). Notably, in no instance were both organisms found in a single sputum sample. Pyrosequencing further confirmed this observation with the number of read numbers of taxa identified. When one species was present, the other did not contribute >1.5% of the total bacterial community. Patients with H. influenzae sputum positivity had a forced expiratory volume in 1 s (FEV1) that was significantly higher than that of those with P. aeruginosa (64.5% versus 48.5%, p<0.01). Clinically, this may be relevant as a form of antagonism between these pathogens, demonstrating an inability to be part of a single core microbiome. Furthermore, in vitro studies have shown that P. aeruginosa inhibits the growth of H. influenzae, suggesting competition within the lung ecological niche [36]. Similar relationships have been observed in paediatric CF patients [37], with speculation that antibiotic pressure and/or patient-specific factors lead to divergence in airway communities. Among all samples, Streptococcaceae, Pseudomonadaceae and Pasteurellaceae were dominant. The authors [35] used soft-class modelling from pyrosequencing data to delineate those patients whose samples were obtained at the time of exacerbation (n=20) from those whose samples were obtained at a time of clinical stability (n=50). Eight individuals from the exacerbation group (40%) had community structures distinct from the remaining patients, with 27 taxa positively correlated with exacerbation, including Burkholderiales, Pasteurellaceae, Streptococcaceae, Xanthomonadaceae, Prevotellaceae and Veillonellaceae. Conversely, 11 taxa were associated with the stable clinical state, including Pseudomonas, Neisseria and Enterobacteriales. Finally, among individuals with frequent exacerbations (defined as more than three in a 12-month period), a subset (37%) could be stratified from infrequent exacerbators between stable and frequent exacerbations. Moraxellaceae, Xanthomonadaceae, Rhodobacteraceae and Staphylococcaceae were positively associated with frequent exacerbation with Campylobacteraceae, Carnobacteriaceae, Corynebacteriaceae, Micrococcaceae, Neisseriaceae and Nocardiaceae positively associated with stability [35]. These observations suggest that bacterial communities of patients with bronchiectasis exacerbations may be more dynamic than those seen in stable patients; however, these observations were not broadly applicable to all subgroups of patients [35].
Rogers et al. [38] aimed to elucidate prognostication by stratifying patient microbiomes in non-CF bronchiectasis using culture (107 adults) and pyrosequencing (96 samples) methodologies. Broadly speaking, three groups were defined among these patients: those dominated by P. aeruginosa (n=26), those dominated by H. influenzae (n=34) and those dominated by other taxa (n=36). Among patients with P. aeruginosa- or H. influenzae-dominated communities, adverse clinical parameters were observed, including significantly worse lung function, higher serum levels of C-reactive protein and higher sputum levels of interleukin (IL)-8 and IL-1b. Predominance of P. aeruginosa, followed by Veillonella species, was the best predictor of future exacerbation frequency, while patients with H. influenzae-dominated communities had significantly fewer episodes. Detection of P. aeruginosa was associated with poor lung function and higher exacerbation frequency, in contrast to the prior study. Taken together, these studies suggest that a “shift” in the microbiome may contribute to clinical exacerbations, with greater granularity observed using culture-independent techniques. However, no clear causative relationships have been discerned thus far. As noted above, colonisation of the lungs by P. aeruginosa is generally exclusive of H. influenzae (and vice versa) and they are rarely co-isolated from sputum, suggesting the potential for strong inter-species competition and inhibitive activity in the lung, which may account for differing abundance profiles in studies [35].
As noted above, abundance does not necessarily correlate with clinical status and patient health as a predictor of clinical stability and pulmonary exacerbations. Less abundant taxa, including Rhodobacteraceae, Xanthomonadaceae, Staphylococcaceae and Moraxellaceae, have been associated with frequent exacerbations [35]. Reduced abundance of satellite taxa, including Streptococcaceae, Prevotellaceae, Veillonellaceae and Actinomycetaceae, has been associated with a decline in the lung clearance index and increased inflammation in patients [39]. The perceived shift in the microbiome may be in part due to increased use of eradication antibiotic therapy for “typical” pathogens, greater use of chronic suppressive antibiotic therapy, increased sputum surveillance and more sensitive molecular techniques for microbial identification [40]. The clinical impact of the microbiome is relevant because bacterial diversity has been shown to significantly positively correlate with FEV1, with bacterial community composition similarly positively correlated with FEV1, neutrophil count and Leicester cough score [31].
The respiratory microbiome and NTM: who is involved?
Several studies have focused on NTM in CF; however, the focus of this review will primarily be on other disease states with microbiome assessment. The reader is referred to several key papers on the former for further reference [41–43]. Identifying the role of the microbiome in NTM disease may provide insight into the pathogenesis and management of the disease, although available studies are limited. Complicating this, there is a lack of consensus around positive isolation of NTM as a coloniser or as a marker of infection, the latter involving direct tissue involvement. Since the discovery of NTM disease, controversy has existed around the isolation of both intermittent and persistent NTM as sequelae of infection. Historical data suggest certain members of the NTM, e.g. Mycobacterium gordonae, Mycobacterium fortuitum and Mycobacterium chelonae, may be seen as the result of long-term colonisation of lungs without apparent disease association [44]. The 2007 ATS guidelines [4] highlight the acquisition of NTM without fulminant disease, particularly in patients with chronic respiratory disease, as potential colonisation and often associated with MAC [45, 46]. However, given the limited understanding of the pathophysiology of NTM and their slow-growing nature, it is unclear if these organisms represent indolent growth as opposed to benign colonisation. Our current understanding of NTM is that colonisation without tissue invasion (i.e. infection) is unknown. Early work by Macovei et al. [47] sought to evaluate the mycobacteriome of healthy humans to elucidate if colonisation does indeed exist. 10 healthy subjects were screened using oral cavity (buccal mucosa and dental plaque) and upper respiratory tract (nostrils and oropharynx) samples with 16S rRNA V3–V4 regions with NTM DNA found in the nostrils of all subjects, and in abundance within the buccal mucosa (n=8), oropharynx (n=7) and dental plaques (n=5). Taken together, these results reveal that healthy individuals may harbour a “nontuberculous mycobacteriome” in their oral cavity and upper respiratory tract. This has implications for our understanding of the balance of colonisation and infection in NTM, in particular within lower respiratory tract communities.
Further unique technical challenges exist in performing comprehensive microbiome studies with NTM. Owing to the slow-growing nature and fastidious culture requirements of mycobacteria, growth may be difficult using standard techniques. Genotypic methods such as DNA sequence analysis of the 16S rRNA gene are often adequate to identify NTM, although some isolates cannot be differentiated with clear granularity at the species level [4]. DNA extraction may be difficult owing to recalcitrant cell composition and slow colony growth, often requiring extra lytic steps in the laboratory pipeline that may affect efficiency and cost [48]. Finally, NTM usually contain only one or two 16S rRNA genes per genome, and thus may be underrepresented among other taxa with more copies of ribosomal genes using standard 16S rRNA sequencing [49]. Consequently, several studies have observed a lack of mycobacteria identification by 16S rRNA gene sequencing in samples with positive cultures for this organism [41, 47, 49]. Refinement of molecular methodology, such as targeting Mycobacterium-specific regions containing the V4 region of the 16S rRNA gene in a nested amplification approach, has been shown to improve identification yield in BAL (47% of BAL samples with NTM culture growth as compared to 27% using standard 16S sequencing [41]).
With the above limitations noted, the microbiomes of patients with NTM do differ from those with NTM-negative samples in several disease states, as summarised in table 2. In 2015, Yamasaki et al. [50] performed the first NTM microbiome study using DNA extraction analysis on BAL samples from patients with non-CF bronchiectasis (n=29) with NTM organisms identified on culture or diagnosed with NTM disease based on ATS criteria. The rates of Haemophilus, Pseudomonas and Staphylococcus abundance were lower while that of Streptococcus was higher in NTM cases compared to controls. Anaerobic species, primarily Prevotella, Fusobacterium, Propionibacterium and Veillonella, were significantly more abundant in NTM patients than in controls (47% versus 18% of the community). On a metabolic level, the higher prevalence of anaerobes may be attributed to mycobacteria inducing local tissue hypoxia, which aids growth [50].
Summary of current studies examining the lung microbiome in individuals with NTM disease#
Sulaiman et al. [49] obtained oral wash and induced sputum samples in 106 non-CF bronchiectasis patients over a 2-year period, with lower respiratory tract samples obtained by bronchoscopy from a subset (n=20) of patients. Using conventional culture techniques, 61 patients (58%) had positive NTM cultures at baseline. In general, sputum samples were enriched with Prevotella, Veillonella and Corynebacterium, while oral wash samples were enriched with Streptococcus, Rothia and Actinomyces species. There was no significant difference in α-diversity (the diversity of the microbiome within one sample) or β-diversity (the diversity of microbial communities between samples) in induced sputum from NTM-positive or NTM-negative patients; however, there was a statistically significant increase in β-diversity among oral wash specimens from NTM-positive patients. Among patients who underwent bronchoscopy, there were no significant differences in the bacterial load, or in α- or β-diversity of NTM-positive versus NTM-negative lower airway samples. BAL samples from NTM-positive participants were enriched with Oxalobacteraceae, while BAL samples from NTM-negative participants were enriched with Porphyromonas [49]. Taken together, this study highlights several critical key difficulties in NTM microbiome studies. First, changes in microbiota between NTM-positive or -negative patients in sputum samples were negligible. By contrast, community differences could be discerned in NTM-positive and -negative patients between upper and lower airway samples from bronchoscopy, suggesting that induced sputum may be a poor representation of lower airway microbiota in this patient population. Second, Mycobacterium were not identified in a large portion of samples with NTM culture positivity using culture-independent techniques, again demonstrating the limited sensitivity of current universal sequencing methodology to study organisms in low abundance, such as NTM [49]. A limitation of this study is the enrolment of patients with mild NTM and primarily MAC subset. It is possible that various strains of NTM, such as more virulent ones including M. abscesses, and greater disease severity may result in unique and robust airway microbiota signatures.
While the majority of the NTM are found in chronic pulmonary disease states, the presence of pulmonary NTM disease has been observed in underlying non-pulmonary medical co-morbidities, as was seen in a small study assessing women with a current diagnosis or history of breast cancer. Philley et al. [51] compared the microbiome of sputa in healthy control individuals (HC), individuals with breast cancer and NTM disease (NTM-BC) and individuals with NTM disease (here referred to as NTM-0). They identified that individuals in the NTM-0 and NTM-BC groups had dominating genera of Haemophilus, Streptococcus, Neisseria, Veillonella, Rothia, Fusobacterium, Leptotrichia and Prevotella whereas the dominating genera for the HCs were significantly different, with enrichment of Streptococcus, Veillonella, Haemophilus, Rothia, Neisseria, Leptotrichia, Prevotella and Fusobacterium. Interestingly, there was no considerable significant difference in genera distribution between the NTM-0 and NTM-BC groups, but the most common top phyla differed between NTM-BC (Bacteroides (36%), Firmicutes (33%) and Proteobacteria (19%)) and NTM-0 (Proteobacteria (28%), Firmicutes (27%) and Bacteroides (13%)) groups. While this study was limited by a small numbers of subjects, a trend towards a difference in the taxa distribution of these groups was identified. The investigators identified a decrease in Veillonella in the NTM-0 group compared to the HC group; this organism is commonly associated with diversity and healthy microbiomes. Fusobacteria sp. were found in significantly higher abundance in the NTM-BC groups, and between those with NTM compared to HCs. This may be clinically significant because certain bacteria, such as Fusobacterium, a pathogenic oropharyngeal genus identified in periodontal and gingival disease, have been associated with malignant states such as colorectal cancer. Finally, there was a significant reduction in α-diversity in the NTM-BC and NTM-0 groups compared to HCs [51]. The study postulates on the functional link between oestrogen and changes in the microbiome, where oestrogen-like compounds can be metabolised to an active form by specific microbiota to promote growth and proliferation of certain microbes, including Bacteroides, Faecalibacterium, Alistipes, Fusobacterium, Prevotella, Staphylococcus and Streptococcus. This increase in oestrogen metabolite is associated with microbial diversity compared to the parent oestrogen, with a potential link to increased risk of breast cancer. The microbial changes observed in this study are not unique to NTM-BC patients, and thus further functional assessment is required to truly elucidate a unique profile in malignancy [51].
The mycobacteriome may play an important role in the balance between NTM disease pathogenesis and healthy states by attenuating bacterial diversity, which has been identified as a protective barrier. As described above, a key limitation to NTM microbiome studies is the reliance on isolation by culture-based techniques given the known limitations of molecular techniques with universal 16S rRNA sequencing for these organisms. A novel study by Cowman et al. [52] enrolled 42 patients with NTM (n=31) and either structural lung disease (non-CF bronchiectasis or chronic obstructive pulmonary disease (COPD)) or no underlying lung disease prior to their diagnosis of NTM disease. Patients were then compared to controls (n=11) with non-CF bronchiectasis or COPD without NTM disease. Molecular identification of NTM was done using the mycobacterial hsp65 gene, a target chosen based on the ability to discern identification at the subspecies level between several key NTM species that may otherwise be identical by partial 16S rRNA sequencing (such as M. abscessus and M. chelonae [53]; and M. abscessus spp. abscessus and ssp. bolletii/massiliense [54]). Using both mock communities and patient samples, mycobacterial sequences were recovered from all samples in both cases and controls, regardless of growth in culture or presence of clinical disease. There was a significant reduction in mycobacterial species diversity (as measured by Shannon diversity index) and species evenness (as measured by Pielou's evenness index) in NTM cases compared to controls. Among NTM cases alone, Shannon index and observed richness were higher in those receiving treatment for NTM, and Shannon and Pielou indices correlated positively with FEV1 [52], suggesting improvement in tangible clinical outcomes with treatment. This study highlights that isolation may indeed be true recovery of NTM, rather than transient inhaled passengers, and sputum culture may indeed lack sensitivity to the presence of NTM. Further, this study used next-generation sequencing with mycobacterial hsp65 directly from sputum samples to provide a profile of mycobacterial communities in the lung, which may serve as an attractive target going forward for broader microbiome work in NTM.
Taken together, NTM microbiome studies suggest a unique community within these patients; however, further analysis needs to be undertaken to explore causative relationships apart from correlational analysis. Cofounding these assessments is the heterogeneic NTM disease patient population, which makes it difficult to apply general conclusions about the microbial communities across diverse diseases. A key limitation highlighted by molecular assessment is that detection of microbial DNA may not necessarily be indicative of viable organisms and thus true clinical implications are unknown. Further studies in subjects with other chronic lung disease apart from bronchiectasis, a known structural risk factor for acquisition of NTM, and healthy individuals will be required to further explore this. Finally, a limitation of NTM microbiome work is the recovery of large amounts of non-mycobacterial sequences, which underpowers the depth of sequencing resolution, and failure to detect NTM sequences at low copies. These limitations may mean that the true community diversity in these patients is under-represented.
Pathogenesis of the mycobacteriome
Unlike M. tuberculosis, which has been the focus of many studies surrounding the mycobacterial immune response, there have been few studies assessing the host response to pulmonary NTM infection and subsequent disease. While there are some similarities between these organisms, differences in NTM include lack of latent state, mode of acquisition and reduced virulence profile [55].
T-helper 1 (Th1) cells, interferon-γ (IFN-γ), tumour necrosis factor-α (TNF-α) and IL-2 have been identified as important components of the host response in NTM disease [56]. Studies in murine models demonstrate that deficiencies in these components lead to a reduced ability to control M. avium infections [56]. However, there is no single immune defect in patients that can be attributed to patients with NTM disease, suggesting that a variety of deficiencies may contribute to the predisposition to NTM. Normal immune responses are often insufficient to effectively clear a mycobacterial infection owing to the organism's ability to evade the immune system [57]. There is further evidence that patients with NTM-related bronchiectasis have a distinct immune phenotype, including increased IFN-γ, TNF-α and IL-12 production, compared to healthy controls; this results in cytokine dysregulation and inability of the host to resist mycobacterial infection and subsequent infection [58, 59]. Lai et al. [60] demonstrated that IFN-γ production by natural killer (NK) cells activates the immune response, as identified by M. kansasii infection in NK cell- and IFN-γ-deficient mice. Depletion of NK cells led to an increased bacterial load and mortality in mice, likely due to a reduction in macrophage phagocytosis and cytokine production. IFN-γ deficient mice exhibited similar results, with injection of recombinant IFN-γ and NK cells into these mice attenuating NTM pathogenesis [60]. Human studies of healthy bronchial epithelial cells identify that components of mycobacterial cell walls, including peptidoglycans and the mycolic acid layer, promote upregulation of pro-inflammatory genes related to type 1 IFN-γ and IL-36. Transcription of these genes also leads to the production of CCL5 and CCL22 chemokines, important leukocyte chemoattractants in disease [61]. Finally, investigating the transcriptional response of epithelial cells during NTM infection has identified downregulation of ciliary genes and upregulation of cytokines and chemokines specifically related to IL-32 and cholesterol biosynthesis, thereby promoting adherence and pathogenesis [62].
How might the microbiome impact these host–microbe dynamics related to NTM? In their study, Sulaiman et al. [49] identified an association between microbial signatures in NTM disease and a distinct mucosal immune phenotype. In NTM-positive patients, BAL samples from affected lung segments had significantly higher numbers of neutrophils and fewer macrophages when compared to non-involved lung segments. In vivo cytokine levels measured in BAL showed a different inflammatory profile for NTM-positive patients, with significantly higher levels of IFN-γ, IL-8, IL-12p70, IFN-γ-inducible T-cell α chemoattractant, macrophage inflammatory protein (MIP)1α and MIP1Bβ. Microbiome signatures associated with inflammatory biomarkers in NTM-positive and NTM-negative samples were also compared. In BAL samples from NTM-positive participants, oral commensals including Prevotella, Veillonella and Leptotrichia co-occurred and demonstrated significant correlations with the number of neutrophils and several cytokines, including IL-6, IL-17, IL-23 and C-X3-C motif chemokine ligand 1. In BAL samples from NTM-negative participants, the relative abundance of oral commensals in the lower airway samples had fewer significant correlations with levels of cytokines. It has previously been shown that IFN-γ induction enhances macrophage activity in mycobacterial infection, leading to the production of cytokines such as TNF-α, necessary for resistance to Mycobacterium infection [63, 64]. In NTM-positive samples, BAL cells stimulated with lipopolysaccharide (LPS) showed significantly blunted IFN-γ and granulocyte–macrophage colony-stimulating factor (GM-CSF) levels, suggesting important impaired innate immune responses. In a co-occurrence network analysis, significant associations between taxa identified as oral commensals, e.g. Prevotella, Veillonella and Leptotrichia, and Th17 cytokines were also seen in NTM-positive BAL samples. Finally, the relative abundance of mycobacteria was not significantly correlated with levels of inflammatory biomarkers, suggesting the importance of inflammatory modulation by other microbes on lower airway inflammation in NTM disease.
With the increase in availability of shotgun sequencing, genomic analysis of NTM species has identified an array of virulence factors designed to facilitate infection and growth within a variety of niches, ranging from soil and aquatic environments to human epithelial biofilms. Genome comparisons to M. tuberculosis using phylogenetic analysis has highlighted the evolution of virulence mechanisms including specific secretion pathways (ESX or Type VII secretion) as well as numerous lipids derived from M. tuberculosis [65]. Interestingly, although many NTM are considered to be opportunistic pathogens, M. vaccae is unique in that it is a transient human coloniser with mutual ecologic benefit to the host. M. vaccae inhibits pulmonary allergic inflammation in mice [66] and decreases anxiety in both mice [67] and humans [68] by a presumed gut–brain–microbiota axis.
The potential interplay between host, microbiome and NTM is complex (figure 1). Postulated mechanisms based on limited available studies suggest unique inflammatory phenotypes in NTM patients. The NTM microbiome study by Sulaiman et al. [49] was the first to discern associations between lower airway microbiota signatures and inflammatory biomarkers; however, larger scale studies in times of clinical stability and acute exacerbation are required to delineate causal relationships. Furthermore, the impact of the microbiome on inflammation in pulmonary disease is an important contributor to this complex ecological milieu. Early work from the Bronchiectasis and Low-dose Erythromycin Study (BLESS) discerned the profile of bronchiectasis patients with more (Pseudomonas- or Veillonella-dominated microbiome) or fewer (Haemophilus-dominated microbiome) exacerbations [38]. Inflammatory profiles were correlated, demonstrating H. influenza-dominated microbiomes with higher levels of matrix metalloproteinases (MMPs) than the frequent exacerbator Pseudomonas-dominated group [38, 69]. MMPs are proteolytic enzymes that break down extracellular matrix as a primary structural component of the lung. They are hypothesised to mediate an element of clinical deterioration in bronchiectasis. Interestingly, while patients with Pseudomonas-dominated microbiomes seemingly have poorer clinical outcomes such as lung function, patients here did not demonstrate elevations in either levels or activity of any MMP when compared to H. influenzae-dominated profiles. The authors speculate this may be due to several mechanisms, including less neutrophil-specific airway inflammation by H. influenzae and subsequent higher levels of MMP secretion from other cells types. Alternatively, Pseudomonas-dominated communities may show reduced MMP secretion owing to decreased viable airway epithelium secondary to more severe disease phenotypes when compared to the former [69]. Taken together, these studies highlight an association between oral commensals and host inflammatory responses, suggesting an unknown modulating effect of other microbiota in NTM lung disease that requires further study [70].
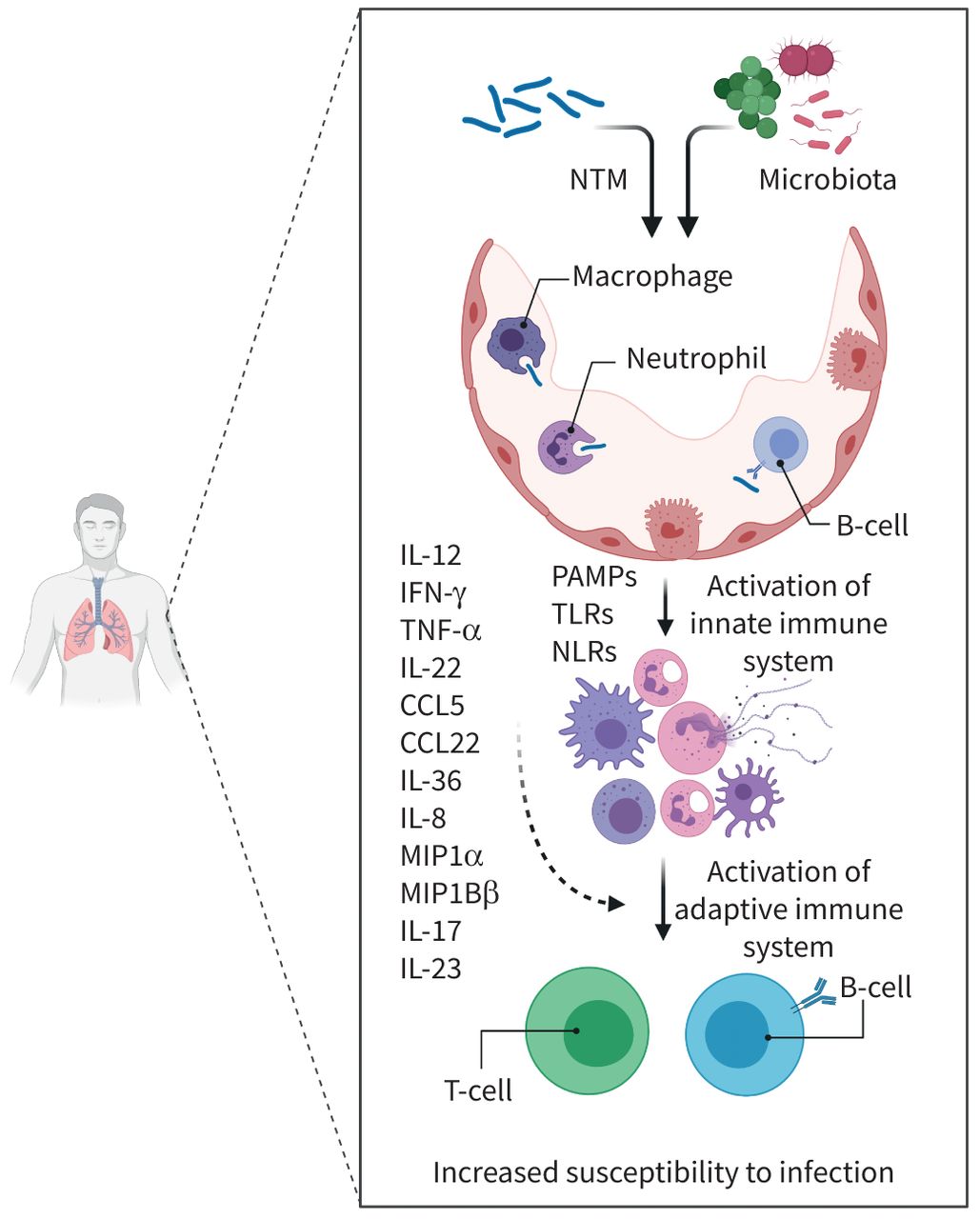
Interactions between nontuberculous mycobacteria (NTM), the microbiome and the host inflammatory response. Upon presentation to the host cell, NTM along with resident lung microbiota induce complex inflammatory interactions that propagate NTM infection and disease. Initial stimulation by the innate immune response is composed mainly of neutrophils, macrophages, dendritic and natural killer cells. Activation is mediated by recognition of mycobacterial and microbial pathogen-associated molecular patterns (PAMPs) through Toll-like receptors (TLRs) and Nod-like receptors (NLRs). Phagocytosis and pathogenesis ensues with stimulation of the cellular-mediated immune response (T-cells and B-cells) through complex cytokine interactions, primarily interferon-γ (IFN-γ), interleukin (IL)-2, IL-12 and tumour necrosis factor-α (TNF-α). Various other cytokine and chemoattract ligands have been implicated in pathogenesis as listed. The role of the microbiome within this inflammatory cascade is complex and intricate within all of the aforementioned pathways. Created with BioRender.com. CCL: chemokine (C-C motif) ligand; MIP: macrophage inflammatory protein.
Effect of NTM antibiotic treatment on the microbiome
Antibiotics are a major cause of microbiota changes because these drugs are designed to attenuate bacterial growth and subsequently cause death. In gut and pulmonary microbiome studies, early antibiotic exposure in childhood has been identified as a risk factor for the development of obesity [71], asthma [72] and diabetes [73]. In adulthood, chronic exposure to antibiotics correlates with microbiome shifts that result in susceptibility towards pathogen colonisation, impaired host responses and chronic microbial dysbiosis [74]. Increasingly intensive antibiotic use in chronic disease states may be a contributory factor to the increasing prevalence of NTM by modulating the existing microbial community [75].
Treatment of NTM lung disease is difficult and involves prolonged drug regimens that are associated with numerous potential side effects and costs [76]. Up to 49% of NTM patients are left with chronically progressive or recurrent disease, requiring an average of 1–2 years of therapy with significant adverse side effects [77]. Guidelines recommend continuing multidrug treatment for 12 months following negative sputum conversion, with relapse or reinfection rates varying from 25% to 50% [16, 57]. Several intrinsic traits of NTM contribute to treatment failure and challenges to the development of new antimycobacterial drugs, including hydrophobic cells due to a lipid-rich impermeable outer membrane, slow-growth, predilection for biofilm formation and clonal variants within the same species conferring differing minimum inhibitory concentration profiles [1, 78]. Currently, guideline-based first-line treatment includes a newer-generation macrolide, ethambutol and a rifamycin (rifampin or rifabutin) [19].
Macrolide and rifamycin drugs demonstrate broad-spectrum activity against a wide range of Gram-positive and Gram-negative bacteria, while ethambutol specifically targets mycobacterial species. The effects of NTM treatment on the intestinal or pulmonary microbiome are unknown. In TB literature, studies in mice and TB patients have examined the effects of treatment in acute and chronic states on the microbiome [74]. These studies demonstrated that conventional TB therapy (including the use of rifamycin and ethambutol) cause a defined and persistent dysbiosis in the intestinal microbiota [79]. Specifically, in combination therapy, rifampin was associated with decreased intestinal diversity and an increased number of Clostridium species in both humans and mice. In mice, monotherapy was analysed and rifamycin was the major driver of taxonomic alterations. In similar experiments, mycobacterial specific therapy including ethambutol also independently affected the microbiota and these changes were distinct from those observed in combination therapy [79]. Macrolides have had success not only as antimicrobial drugs but also as immunomodulatory agents, with treatment across a broad-spectrum of chronic lung diseases including CF, bronchiectasis, COPD and bronchiolitis obliterans. Azithromycin, the preferred first-line macrolide for NTM disease, has been the subject of proof-of-principle studies to discern the impact on the microbiome. Effects of azithromycin have been postulated to span the host and microbiome at multiple sites, including lungs, gut and systemic immunity. Using BAL specimens from emphysema patients, Segal et al. [80] characterised cellular and humoral host response, confirming that inflammatory changes in the lung are dampened in those receiving azithromycin. Host-associated and bacterial-associated metabolites were altered following azithromycin therapy and these subsequent metabolites can blunt alveolar macrophage response to exogenous endotoxin challenges. Further, azithromycin influences systemic inflammation by direct effects on host immune cells and indirect modulation of the gut microbiota, feeding back into lung immunity. Chronic macrolide exposure, such as noted in the BLESS trial with twice-daily erythromycin in patients with bronchiectasis, causes longitudinal chronic changes in the respiratory microbiome, with displacement of H. influenzae by more macrolide-tolerant pathogens including P. aeruginosa [81]. Finally, in in vitro studies, azithromycin affects neutrophil chemotaxis, reduces pro-inflammatory cytokines and chemokines IL-8, IL-1β and NF-κβ, and downregulates the Th2 inflammatory cascade [82].
Despite adherence to guideline-based therapy, the success rate, defined as sputum culture conversion to negative that persists throughout follow-up, may be as low as 53–66% with macrolide-containing regimens [83, 84]. This has prompted the use of newer antimicrobial treatments including amikacin liposoma inhalation solution, clofazimine and bedaquiline. Currently, there are novel agents such as inhaled GM-CSF and phage therapy in the drug development pipeline [57]. The impact of these newer agents on the gut and pulmonary microbiota is unknown and further longitudinal assessment is necessary to evaluate acute and chronic perturbations in the microbial community.
Antibiotic resistance in NTM, in particular in M. abscessus, is a major clinical concern. Intrinsic and acquired mechanisms of antibiotic-resistance genes such as 16S rRNA and 23S rRNA point mutations (rrl and rrs respectively, conferring macrolide resistance), ermB (macrolide resistance) and polymorphisms in gyrA and gyrB (quinolone resistance) are increasing in prevalence [85]. The respiratory habitat and surrounding microbiome of NTM can bring it close to highly virulent pathogens (such as P. aeruginosa in CF [86]) and may serve within the genetic milieu to donate novel drug-resistance molecular mechanisms or virulence factors. Constant ecological selection with antibiotic pressure forces the microbiome to better adapt to external pressures by acquiring antibiotic-resistance genes through horizontal gene transfer, mobile genetic elements and bacterial toxin–antitoxin systems. The impact of this “antibiotic resistome” and the possibility of gene recombination within this vast community may be major contributors to treatment failure in already difficult-to-treat organisms such as NTM.
Future directions and challenges
Conventional culture techniques to investigate microbial infection are the gold standard in the study of infectious diseases, despite suggestions that 90% of microbial community members cannot be cultured with standard methodology [87]. Characterisation of the human microbiota primarily focuses on disease-specific conditions that affect the progression of clinical disease. Few studies have been conducted to explore the microbial diversity in NTM patients compared to healthy controls, in part owing to technical difficulties compared to other microbiome studies. It has been stated that “the use of precision medicine will be the most impactful change in clinical medicine in the next 10 years”, emphasising the simplistic nature of the current one-size-fits-all model of care [88]. Identifying the role of the microbiome in NTM disease pathogenesis using non-culture-based methods may lead to improved management of the disorder [41].
In human studies, the functional microbiome is composed not only of microbes but also a “resistome” (encompassing all antibiotic-resistance genes) and a “virulome” (set of genes encoding virulence). The clinical implications are immense because these bacterial virulence factors may be easily transferred between populations through horizontal gene transfer, allowing commensal bacteria to participate as potential opportunistic pathogens [89]. An important, but unanswered, question is whether differences in the microbiome and host immune system may identify individuals who are susceptible to NTM disease. Evaluation of the lung microbiome in various disease states has identified changes in microbial diversity and/or enrichment with both pathogenic and commensal bacteria. The host may alter the lung microbiome through priming of the immune system, with consequences that may provide a beneficial or harmful response to otherwise “innocent” bystanders. The microbial perspective raises the question of whether pathogenic features may be transferred within a niche environment to mycobacteria and, conversely, whether there are protective factors that may be transmitted (figure 2).

{kind=link}
{kind=link}
Potential mechanistic pathways between nontuberculous mycobacteria (NTM) and the host lung microbiome. The complex interplay between healthy state and dysbiosis is influenced by microbial factors (species present in the microbiome, presence of virulence genes and acquisition of genes by horizontal gene transfer) and host factors (innate and adaptive immune system, external pressure selection such as by antimicrobials). This dynamic ecological niche plays a critical role in the transition between acquisition to infection and finally infection of organisms, such as NTM. Created with BioRender.com.
Characterisation of the lung microbiome in NTM infection may provide novel therapeutic avenues, such as the use of probiotics, prebiotics, antibiotics, enhancement of “good” members of the community or potential elimination of detrimental members by modification of microbial composition [25]. NTM disease is unique because antimicrobial treatment is long, arduous and has variable success. Characterisation of the airway microbiome may be used not only to aid in antimicrobial drug selection, but also as a marker of the effect of therapy by identifying changes in community diversity over time as an early biomarker to determine treatment efficacy. Longitudinal prospective studies with successive sputum samples are required to better elucidate changes in the microbiome composition during NTM infection, disease progression and treatment course. Furthermore, longitudinal studies as compared to cross-sectional analysis may allow identification of dynamic changes to aid in clinical decision management. This will allow further characterisation of the resilience and responsiveness to the community with clinical changes over time. Future research directed towards the role and impact of the respiratory microbiome in NTM incidence and disease progression is critical to fully elucidate host–microbe interactions and to improve patient care.
Footnotes
Provenance: Submitted article, peer reviewed.
Conflict of interest: C.S. Thornton has nothing to disclose.
Conflict of interest: M. Mellett has nothing to disclose.
Conflict of interest: J. Jarand has nothing to disclose.
Conflict of interest: L. Barss has nothing to disclose.
Conflict of interest: S.K. Field reports grants and personal fees from GSK, Novartis and Boehringer-Ingelheim; grants from InsMed; and personal fees from Teva, Canadian Agency for Drugs and Health Technologies (CADTH), and Grifols, outside the submitted work.
Conflict of interest: D.A. Fisher has nothing to disclose.
- Received September 16, 2020.
- Accepted March 9, 2021.
- Copyright ©The authors 2021
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References











Jump To
- Article
- Abstract
- Abstract
- Introduction
- Nontuberculous mycobacteria
- The respiratory microbiome and the landscape of microbial players
- The respiratory microbiome and NTM: who is involved?
- Pathogenesis of the mycobacteriome
- Effect of NTM antibiotic treatment on the microbiome
- Future directions and challenges
- Footnotes
- References
- Figures & Data
- Info & Metrics