





Abstract
Idiopathic pulmonary fibrosis (IPF) is one of the most common forms of interstitial lung disease presenting in persons 50 years and older. Through a comprehensive review of available studies, we aimed to assess health-related quality of life (HRQoL) of people living with IPF and the instruments used in this assessment.
Searches were conducted up to May, 2020. Quality appraisal and data extraction were performed using pre-designed forms. Narrative synthesis approach was used to report results of the systematic review and a random effects model was used for the meta-analysis. A leave-one-out sensitivity analysis was performed, and a trim and fill method was used to assess publication bias.
The review included 134 studies. The most used instruments to measure HRQoL were St George's Respiratory Questionnaire (SGRQ), Short Form 36 (SF36) and EuroQoL (EQ5D). Standardised mean scores (95% confidence interval) for these instruments were as follows: SGRQ total score: 44.72 (42.21–47.22); SF36 physical component score (PCS): 37.00 (34.74–39.26) SF36 mental component score (MCS): 50.18 (48.41–51.95); King's Brief Interstitial Lung Disease questionnaire total score: 58.38 (55.26–61.51); and EQ5D utility: 0.73 (0.68–0.79). Analysis of standardised means for both SGRQ and SF36 demonstrated worse scores in physical health domains as compared to mental health domains.
This systematic review confirms that IPF negatively affected HRQoL, mostly impacting the physical health domains. This study also demonstrated that a diverse number of instruments are used to evaluate HRQoL. In view of this diversity, a standardised approach to measurement of HRQoL for IPF is important to ensure that comparisons made are reliable.
Abstract
This study established that a wide variety of instruments are used to evaluate HRQoL in IPF and suggests that to improve comparability across studies, a standardised approach to the measurement of HRQoL is important https://bit.ly/2XJSApz
Introduction
Idiopathic pulmonary fibrosis (IPF) is one of the most common interstitial lung diseases in the elderly [1, 2]. It is characterised by progressive fibrosis and destruction of lung tissue, which leads to variably rapid decreases in lung function and eventual respiratory failure and death [3]. Although the disease course may vary, evidence suggests the median survival time for IPF from diagnosis is 2–4 years [4, 5]. People with IPF usually experience increasingly debilitating symptoms including cough, dyspnoea, fatigue and weight loss [6], all of which have an impact on their daily life. Treatment options for the disease are quite limited and only two recently introduced drugs are known to slow disease progression but not cure the disease [7].
Given this combination of symptom burden, poor prognosis and limited treatment options, it is important to ascertain the impact that the disease has on quality of life. How the disease impacts an individual's quality of life or the health-related quality of life (HRQoL) is determined by several factors, including the disease course, the individual's perceptions of their disease and their coping mechanisms [8]. Consequently, a holistic approach to the management and care of people with IPF and the evaluation of new therapies should take into consideration how these therapies improve HRQoL in addition to improving clinical outcomes.
To improve the HRQoL in people with the disease we must first ascertain the best methods to measure it accurately and reliably. To date, research in this field has been limited [9]. In addition, there is still some debate about the appropriate instruments to measure HRQoL specifically in IPF [9, 10]. Several instruments have been used to measure the HRQoL in IPF, including generic instruments such as the St. George's Respiratory Questionnaire (SGRQ), and Short Form-36 (SF36). With novel anti-fibrotic drugs now available and active research ongoing into the development of other drugs and treatment modalities, it is important to establish a standardised approach for the measurement of HRQoL as an outcome of treatment. This will assist in harmonising comparisons made across populations and intervention studies.
In this current study, we conducted a comprehensive review of existing literature to evaluate studies that have examined HRQoL in IPF. We sought to determine how IPF affected HRQoL, and the measurement tools used in this assessment. Moreover, this study expands on previously conducted reviews on HRQoL [11, 12] as many studies have been published over the last 3 years.
Methods
This review was done in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analysis Protocols and registered to the international prospective register of systematic reviews, PROSPERO (CRD42018111292). This protocol was subsequently updated to include a meta-analysis.
Search strategy
Searches were conducted up to May 2020, in MEDLINE, EMBASE, PSYCHINFO, SCOPUS, the Centre for Reviews and Dissemination (CRD), Database of Abstracts of Reviews of Effects (DARE), National Health Service Economic Evaluation Database (NHS EED) and the Cost-Effectiveness Analysis Registry (CEA Registry). Search strategies for the databases are included in the supplementary material (supplementary material S3). Hand searches were conducted using reference lists for the retrieved studies and previous systematic reviews [11, 12] to identify additional studies.
Study selection
Studies were included if they were intervention or observational studies; if they included and reported measurements on quality of life; and if the reporting language was English, French, Spanish or German. Inclusion and exclusion criteria are available in supplementary material S2.
Endnote referencing software [13] was used to catalogue all searches and to omit duplicates. The studies that remained after duplicate removal were screened by two reviewers simultaneously using Covidence online software [14] in two phases: title/abstract and then full text. This was completed by two reviewers simultaneously (I.A. Cox, B. de Graaff). A third reviewer (E.H. Walters) was consulted if there were conflicts in the study selection.
Data extraction and management
Data from the selected studies were extracted into predesigned Excel spreadsheets. The following variables were collected: authors, country of study, publication year, study type, clinical standards for diagnosis, quality-of-life instrument used and quality-of-life scores reported (mean and standard deviation), number of participants with IPF, proportion of males, mean age and forced vital capacity (FVC) as % predicted. For studies that involved an intervention, the quality-of-life measurements taken before intervention commencement were used.
Quality assessment
Quality assessment of the studies was conducted using a combination of criteria adopted from previous studies [6, 12, 15–17]. These evaluated the IPF case definition, demographic and clinical characteristics of the IPF participants, clinical diagnosis standards, the HRQoL measurement instrument selection and the study endpoints (supplementary material S4). A points-based system was used to calculate the total score for each study, which was then represented as a percentage of the highest attainable score. Studies were then classified into low (<50%), medium (50–75%) and high (>75%) quality based on the percentage calculated.
Data analysis
A descriptive approach including use of figures and tables was used for reporting the results of the systematic review. Pooled means were calculated for age and FVC % pred. The mean age was then categorised into age groups (supplementary material S5-1) and FVC % pred. was used to categorise disease severity based on the level pulmonary function impairment [18–20] (supplementary material S5-2). In studies for which standard deviations were not reported for quality-of-life scores, standard errors and sample size were used to calculate the missing data [21]. In studies for which medians were reported for quality-of-life scores, means and standard deviations were calculated using methods by Hozo et al. [22] and Luo et al. [23].
The R software [24] and the “metafor” package [25] was used to conduct the meta-analysis. When multiple papers were reporting on the same study, the paper with the largest sample size was used in the meta-analysis. The between study variability was considered using the random effects model. The I2 statistic was used to evaluate heterogeneity and inconsistency across the studies [26, 27] and the Cochrane Handbook was used to classify the levels of heterogeneity [28]. To further evaluate the cause of heterogeneity, a subgroup analysis was conducted in which two or more studies were available, for disease severity, age group and study quality. Sensitivity analyses were conducted using two methodologies. The first method omitted one study at a time [28, 29]. The second method arranged the studies by sample size in ascending order (smallest to largest), then starting with the three smallest studies, one study was added at a time, to assess the influence on the mean [30]. Finally, the “trim and fill” method was used to assess publication bias [28, 29].
Results
Initial searches identified a total of 3578 abstracts from databases and 15 studies from reference list hand searches. After the screening process, a total of 134 studies conformed with the inclusion criteria and 97 were included in the meta-analyses. The Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram (figure 1) summarises the selection process. All reasons for exclusion of full text studies are explained in the supplementary material S6. All studies included in the meta-analyses are reported in supplementary material S7.
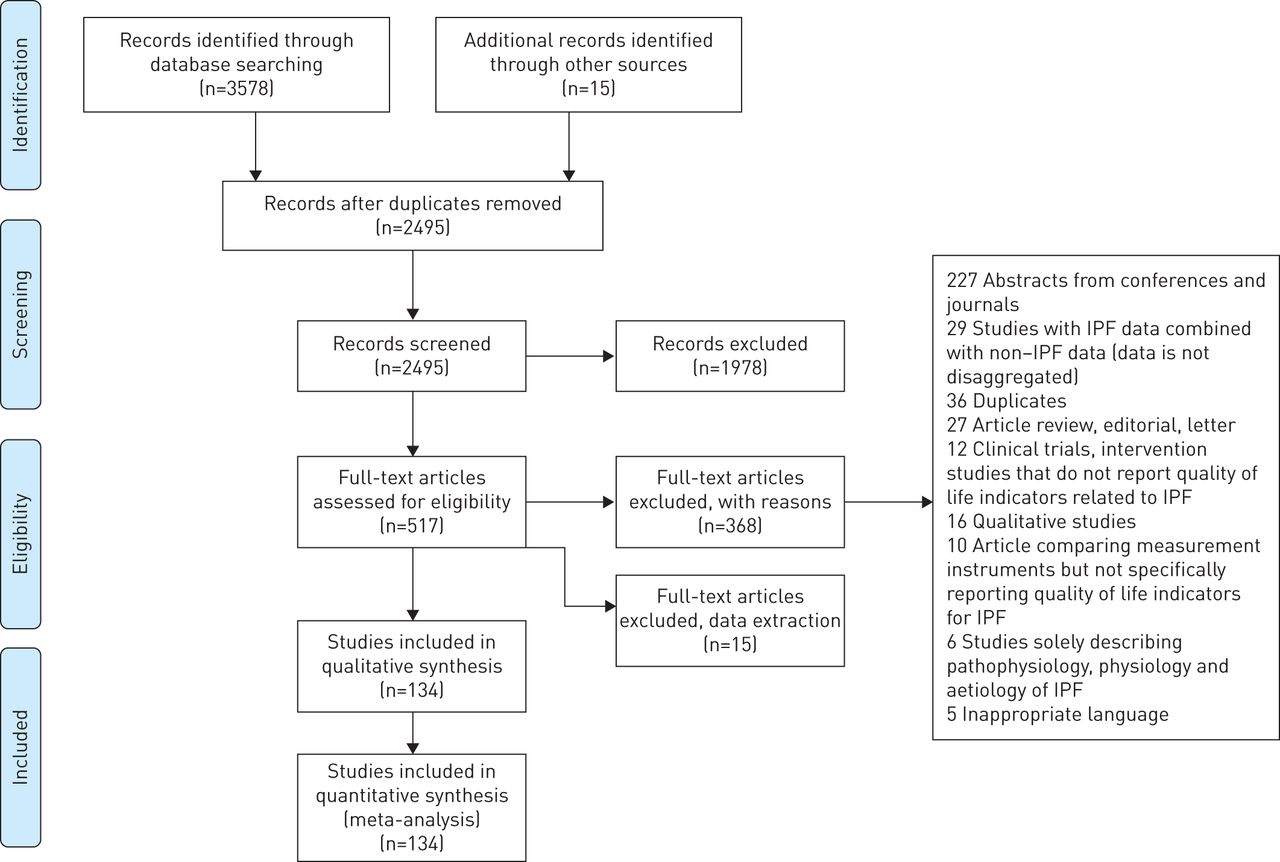
Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow chart illustrating study selection process.
General characteristics of all studies included
The number of studies published has increased substantially in the past 5 years (figure 2). Approximately 57% of all the studies included were published between 2015 and 2020 while the remaining 43% of the articles were published between 2000 and 2014 (figure 2). Most studies were from Europe (33%), North America (23%) and Asia (20%). Multi-country clinical trials accounted for 18% of all studies and the USA (20%) and Japan (13%) were the most studied individual countries. Detailed information on individual studies can be found in table 1.

Number of included studies by publication year.
Summary of all studies included in the review
Participant characteristics
The participants for most studies were predominantly male, excepting for five studies [31–37]. A high proportion of studies (92%) had participants whose mean age was ≥60 years with the exception of 10 studies [33, 34, 38–45]. The remaining studies did not report the age of the participants. Only a small number of studies reported on ethnicity most of whom were Caucasian (table 1 and supplementary material S8).
Clinical diagnosis and staging
Clinical diagnosis in most studies (87%) was based on one of the following international consensus standards: American Thoracic Society (ATS)/European Respiratory Society (ERS) or ATS/ERS/Japanese Respiratory Society (JRS)/Latin American Thoracic Society (ALAT), dependent on the year the study was conducted. In cases where the international guidelines were not used, one study used the Chinese Thoracic Society (CTS) standards [46], another used ICD-10 coding [47], and one relied on participants’ self-reported diagnosis [48]. The criteria used for the remaining studies was not defined [32–34, 36, 39, 40, 42, 43, 46–57].
With reference to disease severity [18–20], 51% of studies reported on participants with moderate disease severity, 37% were on participants with mild disease while only 4% of studies included participants with severe disease [33, 40–42, 55] (table 1 and supplementary material S8).
Study quality
Supplementary material S4 provides a summary of the study quality criteria used and how the studies performed against each individual criterion. Most studies were of medium quality, 28% were of high quality and 6% low quality.
HRQoL measurement instruments
A diverse number of instruments were used in the studies, with predominance of the use of the generic form of the SGRQ either used singularly or combined with other instruments (60%). The SGRQ is a disease-specific instrument for obstructive lung disease with three domains: activity, impact and symptoms; and a total score. The total score for each domain is depicted as a percentage of the overall impairment on a scale of 0–100%, with 0% and 100% being the best and worst possible health status respectively [58].
The SF36 was the second most used instrument (30%). The SF36 is a generic instrument used to measure HRQoL across several disease conditions. It has eight domains: physical functioning, physical role, general health, bodily pain, vitality, social functioning, emotional role and mental health; and two summary scores: physical component score (PCS) and mental component score (MCS). All of these are measured on a scale from 0–100, where lower scores indicate a worse health status [59].
The EQ5D was not used individually in any study but was used in combination with other assessment instruments in 15 studies (15%). Two versions of this instrument were used in the studies reviewed, the EQ5D-3 level and the EQ5D-5 level. This generic multi-attribute instrument is used for measuring HRQoL over several disease conditions. It reports two measures: the visual analogue scale (VAS) which ranges from 0–100 where a higher score reflects better HRQoL; and, secondly, the health state utility value which is used in economic evaluations. The utility score is reported on a scale between 0 and 1, where 1 represents the best quality of life and 0 represents death. Negative scores are possible, indicating states considered worse than death [60].
12 studies made use of the ILD-specific King's Brief Interstitial Lung Disease questionnaire (KBILD) [36, 54, 56, 61–69]. This instrument measures three domains: psychological, breathlessness/activities and chest symptoms. The scores range from 0–100, where 100 represents the best score [36].
A limited number of studies used disease-specific (IPF) instruments, although many of the studies were conducted after their development in 2010. Of these, five studies [52, 70–73] used the disease-specific format of the SGRQ, the SGRQ-I, and three used “A Tool to Assess Quality of life in IPF” (ATAQ-IPF) [44, 48, 74].
Meta-analysis
SGRQ
60 studies were included in the meta-analysis. The pooled mean scores (95% CI) were as follows: activity, 57.13 (48.40–65.87), symptoms, 50.82 (48.32–53.32); impact, 37.00 (34.41–39.68); and total, 44.72 (42.21–47.22). Domain scores reflecting physical wellness (activity and symptoms) were generally worse than those reflecting emotional wellness (impact).
Both disease severity and age group were observed to have an influence on the SGRQ score. The pooled mean score worsened with increasing disease severity. For age group, there was no consistent pattern, but the worst scores were recorded in the youngest age group (figure 3a). The subgroup analysis on quality of the studies demonstrated that there was a minimal difference between the total scores for medium- and high-quality studies. Low-quality studies had a much higher total score, 57.94, signifying a lower quality of life; however, only four studies were included in this analysis and the degree of certainty was low as demonstrated by the wide confidence intervals.

Forest plots showing pooled means and subgroup analysis for St George's Respiratory Questionnaire (SGRQ) and Short Form 36 (SF36). QOL: quality of life.
SF36
32 studies were included in the analysis. Pooled mean scores (95% CI) for the PCS and MCS were 37.00 (34.74–39.26) and 50.18 (48.41–51.95), respectively. All other domain scores are summarised in supplementary material S9. Like the SGRQ, the domains related to mental wellness had better scores than those related to physical wellness.
Once more, disease severity and age group influenced the pooled mean scores for both physical and mental health domains. Thus, HRQoL deteriorated as the disease progressed in severity and worse HRQoL was generally seen in the younger age groups. (figure 3b and 3c). This pattern is consistent with the results of the SGRQ subgroup analysis. Similar to the SGRQ, there was little difference between the summary scores for both physical and mental health domains in the subgroup analysis for study quality.
EQ5D
There were a limited number of studies with results for the EQ5D. Nine studies were used in the utility analysis and 12 studies for the EQ VAS analysis. The pooled mean scores (95% CI) for the utility and EQ VAS were 0.73 (0.68–0.79) and 65.66 (61.75–69.58), respectively.
The power of the subgroup analysis for EQ5D was limited as a result of the low number of studies available (figure 4c)

Forest plots showing pooled means and sub-group analysis for EuroQoL (EQ5D) and King’s Brief Interstitial Lung Disease questionnaire (KBILD). QOL: quality of life.
KBILD
12 studies were used in the analysis for the KBILD. Pooled mean total score (95% CI) was 58.38 (55.26–61.51).
The power of the subgroup analysis for KBILD was also limited as a result of the low number of studies available. However, the total score for the KBILD was negatively impacted by disease severity, as there was a decrease in the total score with increasing disease severity. The younger age group, 60–69 years, had a worse score than the older age group, 70–79 years (figure 4d).
Sensitivity analysis and bias
Sensitivity analyses showed that there were no outliers (figure 5a and supplementary material S10-1). To corroborate our results, we used a second method based on the sample size of the study (figure 5b and supplementary material S10-2). The results again showed little change in the scores for all the instruments except for the KBILD and SGRQ, which demonstrated a minimum score 5.9 and 4.4 points less, respectively, than the calculated mean from the meta-analysis. This, however, is within the minimal important difference for this instrument which is between 6–10 for KBILD and 5–8 for the SGRQ [75, 76].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Summary plots for sensitivity analyses and funnel plot for assessment of publication bias. QOL: quality of life; EQ5D: EuroQoL; SGRQ: St George's Respiratory Questionnaire; SF36: Short Form-36; KBILD: King's Brief Interstitial Lung Disease questionnaire.
The trim and fill method yielded an asymmetrical funnel plot which can be attributed to high between-study heterogeneity (figure 5c).
Discussion
To date this is the first systematic review with a meta-analysis statistically evaluating impacts on HRQoL in IPF patients, and the patient reported outcome measures (PROM) used in IPF studies. Our results show that, although studies have been published from the early 2000s, there has been an influx of research on HRQoL in the last 4 years, and even more specifically since 2017. Furthermore, though disease-specific instruments have been developed, there is still a predilection for the use of generic multi-attribute instruments. The most frequently used instruments for measuring HRQoL were SGRQ, SF36, KBILD and EQ5D. These instruments demonstrated that the impact of IPF is not restricted only to respiratory limitations but to all aspects of life, including physical, emotional and social functioning; however, the most affected aspect was the physical domain. HRQoL is further influenced by age and disease severity. When compared with the general population and COPD patients with moderate disease, HRQoL is substantially worse in people living with moderate IPF. Taken together, the data from this review extend and update our previous knowledge and confirm that IPF markedly affects the HRQoL of people living with the disease and that this can be measured quantitatively.
Our study demonstrated an increase in the number of studies in the past 4 to 5 years. This can be attributed to the increase in studies which have used HRQoL as an outcome measure in clinical trials and other studies [7]. Clinical trials continue to evaluate new drugs and interventions that may halt disease progression and, as a result, extend life and improve quality of life. Currently there is no treatment to cure IPF. In particular, in recent years, two new drugs, pirfenidone and nintedanib have been approved for the treatment of IPF in many countries [7]. These have been shown to improve lung function and slow progression of the disease in people with mild-to-moderate disease, but not to reverse or cure the disease [128, 170]. Given this, there has been an emphasis and increase in research in the past few years to ascertain optimal dosing, optimal timing of therapy and potential combination therapy using these two drugs in addition to research for new therapies [171]. However, there is still some uncertainty as to the utility of using HRQoL as an outcome measure for these studies [172]. In the first instance, there is debate on the most appropriate instrument that should be used [173]. Although some of the most used instruments such as the SGRQ and SF36 have been validated and deemed appropriate for measuring HRQoL in people with IPF, there is still discussion on whether they are sensitive enough to measure disease-specific deficits [8, 172]. Secondly, many of these instruments have not convincingly demonstrated major changes in HRQoL in some of the most important clinical trials [8]. Lastly, there is still some ambiguity on the how the side effects of the drugs under investigation influence the reporting of HRQoL [172].
Our study showed that the three most-used instruments for measuring quality of life in IPF were SGRQ, SF36, KBILD and EQ5D. Finding the appropriate instrument for measurement of HRQoL has been an ongoing debate in the field of IPF [8]. Both the SGRQ and SF36 have been validated and have been shown to have adequate psychometric properties to assess HRQoL in IPF [43, 143, 162] and have been used in clinical trials to measure outcomes. Notwithstanding, consensus on whether they are sufficiently sensitive to capture unique aspects of the disease is undecided [9]. Although this may be accurate, studies have demonstrated that SGRQ and SF36 yield HRQoL scores that correspond to physiological or clinical measures, showing low scores in the domains corresponding to disease-related deficiencies. Peng et al. [88], Nishiyama et al. [79] and Martinez et al. [43] were all able to demonstrate concurrence between clinical and physiological measures and HRQoL measured by the SGRQ and SF36. Although this is beyond the scope of this paper, these two instruments have also been proven to be sensitive enough to identify minimally important changes in clinical status [76]. Swigris et al. [76] demonstrated that both the SGRQ and SF36 were able to detect small improvements or decline in clinical status of study subjects. For the SF36, a change of 2–4 points was associated with changes in the clinical status, whereas with the SGRQ, a change of 5–8 was associated with clinical status change [76]. According to these findings, we can infer that these two instruments are suitable options for use in clinical trials where HRQoL is being measured as an outcome, with the caveat that disease-specific deficiencies may not be captured.
Our study recorded a pooled mean utility of 0.73 (0.68–0.79). To put this into perspective, when we compare this value to a similar cohort of the general population the utility values is 0.825 with a standard error of 0.005 [[0] and with a similar cohort of persons with moderate COPD, the utility value is 0.74 (0.66–0.83) [174], signifying that persons with IPF generally have a poorer HRQoL. This is one of the advantages of multi-attribute instruments like the EQ5D. When used in the assessment of HRQoL of persons living with IPF they enable comparisons with other disease entities or other populations, and allow the generation of health state utility values that can be utilised in the calculation of quality-adjusted life years, an important measure used in reimbursement decision making and health economic evaluations [175]. Despite this advantage, the EQ5D is not one of the instruments recommended for measuring HRQoL for IPF as there is little evidence on its measurement properties in relation to IPF [10]. For this reason, the instrument has been used in combination with other validated generic instruments or disease-specific instruments in studies. Mapping of validated instruments to the EQ5D has also been an option to generate data for economic evaluations. Although this has been done in many disease conditions for the SF36, it has not been done for IPF. Freemantle et al. [176], however, conducted a mapping of SGRQ to the EQ5D in IPF patients, and Swigris et al. [76] conducted a similar mapping exercise for the SGRQ to the SF36. Given the continued research into optimal therapies and the need for economic evaluation to assess the cost effectiveness of these for health systems, it may be appropriate at this point to conduct validation studies for the EQ5D and, in addition, evaluate other multi-attribute instruments, to assess their suitability in comparison with the EQ5D, for assessing HRQoL in people with IPF. The establishment of minimal inhibitory difference for IPF for these instruments is also important if they are to be used in relation to clinical outcomes. These are significant issues and we will seek to address some of these in our future work.
Our analysis of the KBILD, albeit limited by the number of studies, further confirmed that IPF had a detrimental effect on HRQoL. The KBILD initially designed for ILD has shown tremendous promise for assessing HRQoL in IPF, especially in capturing disease-specific deficits. One of the advantages of this instrument is that it is simple enough to be used both in the research and clinical setting [177]. Validation studies have shown that the instrument demonstrates high internal consistency, good validity, good reliability and responsiveness [36, 56, 75, 177] in the assessment of HRQoL of IPF patients. In relation to the responsiveness, recent research has also demonstrated that the instrument is sensitive enough to detect MCIDs with scores as low as 2.9–4.9, which further supports the instruments suitability for use in IPF research and clinical use [36]. Notwithstanding the successes of this instrument, longitudinal studies with larger cohorts are needed; however, this can be quite challenging given the natural history of the disease and short survival times. However registries such as the Swedish IPF registry are a good opportunity for longitudinal validation as it makes use of this instrument [61].
Our review revealed that paradoxically very few studies used disease specific instruments. Psychometric evaluation and validation studies have shown that these tools have considerable potential in capturing disease-specific properties that influence HRQoL, compared with the generic instruments [36, 56, 74, 178]. The ATAQ-IPF and the SGRQ-I are two of the older disease specific instruments which have been validated, but newer instruments continue to show potential. The Living with IPF questionnaire (L-IPF) [179], an updated and improved version of the ATAQ-IPF, based on patient feedback of the former instrument, is currently under validation [180]. The Patient Reported Outcome Measure (IPF-PROM), which was recently validated, has demonstrated good reliability, validity and correlates strongly with other instruments such as the SGRQ and moderately with FVC [181]. This instrument is currently being evaluated for its responsiveness. Disease specific patient reported experience measure questionnaires (PREMs) are a novel opportunity to capture information that will complement PROMs. The IPF-PREM [182] was developed to complement current PROMs and capture information on patient experience with service delivery and IPF-specific medication in the UK. The Patient Experiences and Satisfaction with Medications (PESaM) Questionnaire [183] captures information patient experiences with IPF-specific medication. Both are still undergoing validation but will offer insight into areas that do affect patient outcomes. The prospects that all of these instruments offer are quite encouraging for the future monitoring of patients with IPF; however, further research beyond validation and their responsiveness is needed to assess their suitability in the clinical setting to monitor patients.
Our results demonstrate that having IPF affects all measured aspects of HRQoL with the physical health domains being most affected. Both the SGRQ and the SF36 identified worse deficits in the physical health and functioning domains compared with psychosocial domains. Given the natural history of the disease and the main symptoms, dyspnoea, cough and fatigue [3], the larger deficit in the physical health domains is expected. Albeit that the severity of these symptoms can be measured objectively through diagnostic tests, patient perspectives on how it affects their daily life and coping mechanisms provides a realistic life perspective that should be used to guide patient management and to assess new therapeutic outcomes of ongoing and future research [8]. Both instruments also showed deficits in domains related to emotional functioning, consistent with available evidence that people with chronic diseases are abnormally predisposed to anxiety and depression [8]. Dyspnoea and coughing in IPF have been directly linked to anxiety and the poor prognosis of the disease has been linked to depression [8].
The subgroup analysis of different age groups using the SGRQ, SF36 and KBILD showed worse pooled mean scores in younger age groups, irrespective of disease severity. Although this was not expected, further review of the literature revealed a population-based study in the USA of people with chronic diseases, which demonstrated similar results. The study included 201 711 people with chronic diseases and demonstrated that younger age group reported poorer quality of life [184]. One plausible explanation is that younger age groups may still be employed and more socially active so the limitations imposed by chronic disease may be more restrictively impactive than for older persons. However, this outcome was not seen with the EQ5D. Potentially, the EQ5D may not be sensitive enough to detect disease-specific deficits [54] and, additionally, the instrument has not been validated for ILD and, more specifically, IPF [54]. Secondly, the number of studies used in the synthesis for the EQ5D is small, and as a result of this, there is potential for inaccurate estimation of the in-between study variance and resultant pooled means during the analysis [185]. Lastly, it is important to note that patterns that may be seen in the individual studies may not necessarily translate when the studies are aggregated in a meta-analysis and this may be as a result of aggregation bias [186]. The subgroup analysis for all instruments for disease severity confirmed that disease severity itself had a negative impact on HRQoL. This was demonstrated in particular in the studies by Glaspole et al. [53] and Leonard et al. [187], which compared gender and lung physiology (GAP) stages with quality-of-life measures. Both studies established a correlation between a higher GAP stage and worse HRQoL scores.
There wasn't clear evidence of publication bias; however, it should be noted that when there is a high level of between-study heterogeneity the bias detection tests are less reliable [29]. Nevertheless, the influence of individual studies on the overall meta-analysis for each instrument did not show significant outliers signifying that the results of the analyses were robust and not affected by the bias. Additionally, subgroup analysis was performed to account for the influence of the demographic and quality of the study variability in the studies. Lastly the study excluded abstracts which may introduce bias due to a lack of peer review.
This study improves on previously published reviews [11, 12] as it contains twice as many studies and was able to include a meta-analysis for the first time. There were, however, some limitations to the study. First, HRQoL scores in most studies did not consider comorbidities which are frequent in the age range under study. Although the exact mechanism of how comorbidities affect the disease process in IPF is still unclear, studies have demonstrated that improving the management of comorbidities improves quality of life and survival in patients with IPF [8, 188, 189]. Secondly, the clinical standards for diagnosis of IPF varied, with some studies using standards that were not in accordance with international consensus [46–48] and in cases where international consensus standards were used, the standards for diagnosis varied since there were updates to the standards in 2000, 2011, 2015 and in 2018. In addition to the aforementioned, the exclusion of studies on the basis of language may introduce some bias in the analysis. The consensus on this, however, is that the effect may be minimal [28]. Lastly, we omitted grey literature from our study which may increase publication bias in the analysis [190]. There is, however, a lack of consensus on the effect of the inclusion or exclusion of grey literature on publication bias in systematic reviews and meta-analyses [191].
In conclusion, this study extends previous knowledge by showing that IPF has detrimental effects on HRQoL of people living with the disease and the domain most affected is that measuring physical functioning. Given the demonstrable poor quality of life in addition to the already well-known poor survival time related to IPF patient-management strategies should not only be focussed on clinical outcomes. Emphasis should also be placed on strategies to improve quality of life of people living with the disease with a more holistic approach to care.
Conclusion
This study set out to evaluate HRQoL in IPF and how it is measured. Our review demonstrated that HRQoL is markedly affected by IPF and that the most affected part of life is that related to physical functioning. Furthermore, our review revealed that there are a diverse number of instruments used to measure HRQoL in people with IPF, many of which are not disease specific instruments although they have been validated for the disease. In view of this and emerging research to find optimal therapies to treat the disease, a standardised approach to measure HRQoL is needed. This will allow for a more harmonised approach to comparisons across trials and different populations.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary_material ERR-0154-2020_Supplementary_material
Acknowledgements
The authors would like to acknowledge Petr Otahal of the Statistics Department at Menzies Institute for Medical Research (Tasmania, Australia) for technical statistical support.
Footnotes
Provenance: Submitted article, peer reviewed
This article has supplementary material available from err.ersjournals.com
Author contributions: I.A. Cox designed the study, developed the search strategies, conducted the screening of the titles, abstracts and full texts, interpreted results and wrote the first draft of the manuscript. N. Borchers Arriagada contributed to the design and performance of the statistical analysis and manuscript writing. B. de Graaff contributed to the study design and search strategies, conducted the screening of the titles, abstracts and full texts, and contributed to the manuscript writing. H. Walters contributed to study design, resolution of discrepancies and manuscript writing. S. Lartey performed the data audit and contributed to manuscript writing. T.J. Corte contributed to manuscript writing. I. Glaspole contributed to the manuscript writing. A.J. Palmer contributed to conceptualisation and study design, development of search strategies and manuscript writing. All authors approved of the final version of the report.
Data availability: The datasets generated and analysed during the current study are available from the corresponding author upon reasonable request.
Conflict of interest: I.A. Cox reports grants from the National Health and Medical Research Council, during the conduct of the study.
Conflict of interest: N. Borchers Arriagada has nothing to disclose.
Conflict of interest: B. de Graaff has nothing to disclose.
Conflict of interest: T.J. Corte reports grants and personal fees from Roche and BMS; grants, personal fees, non-financial support and other from Boehringer Ingelheim; grants from Actelion, Bayer, Galapagos and Biogen; personal fees from AstraZeneca; and other from Promedior, outside the submitted work.
Conflict of interest: I. Glaspole reports personal fees from Roche and Boehringer Ingelheim, outside the submitted work.
Conflict of interest: S. Lartey has nothing to disclose.
Conflict of interest: H. Walters has nothing to disclose.
Conflict of interest: A.J. Palmer reports grants from the National Health and Medical Research Council, during the conduct of the study.
Support statement: Funding for this study came from the Centre of Research Excellence in Pulmonary Fibrosis (funded by the National Health & Medical Research Council grant 1116371 and supported by Foundation partner Boehringer Ingelheim and Program partners Roche and Galapagos). Funding information for this article has been deposited with the Crossref Funder Registry.
- Received May 20, 2020.
- Accepted June 3, 2020.
- Copyright ©ERS 2020.
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References









