





Abstract
Monogenic type I interferonopathies are inherited heterogeneous disorders characterised by early onset of systemic and organ specific inflammation, associated with constitutive activation of type I interferons (IFNs). In the last few years, several clinical reports identified the lung as one of the key target organs of IFN-mediated inflammation. The major pulmonary patterns described comprise children's interstitial lung diseases (including diffuse alveolar haemorrhages) and pulmonary arterial hypertension but diagnosis may be challenging. Respiratory symptoms may be either mild or absent at disease onset and variably associated with systemic or organ specific inflammation. In addition, associated extrapulmonary clinical features may precede lung function impairment by years, and patients may display severe/endstage lung involvement, although this may be clinically hidden during the long-term disease course. Conversely, a few cases of atypical severe lung involvement at onset have been reported without clinically manifested extrapulmonary signs. Hence, a multidisciplinary approach involving pulmonologists, paediatricians and rheumatologists should always be considered when a monogenic interferonopathy is suspected. Pulmonologists should also be aware of the main pattern of presentation to allow prompt diagnosis and a targeted therapeutic strategy. In this regard, promising therapeutic strategies rely on Janus kinase-1/2 (JAK-1/2) inhibitors blocking the type I IFN-mediated intracellular cascade.
Abstract
Progressive severe lung impairment may occur clinically hidden during monogenic interferonopathies. Pulmonologists should be aware of the main patterns of presentation in order to allow prompt diagnosis and initiate targeted therapeutic strategy. https://bit.ly/2UeAeLn
Introduction
Monogenic type I interferonopathies comprise a group of inherited heterogeneous disorders characterised by early onset of systemic and organ specific inflammation associated with constitutive activation of type I interferons (IFNs) [1–8]. Since their initial description in 2011 [1], type I interferonopathies have drawn the attention of many parts of the scientific community, and involved expanding medical fields as the understanding of these entities has grown. The concept of the potentially harmful effects of an unrestrained IFN pathway was first outlined by Gresser et al. [9] in rodents. Subsequently, Lebon et al. [10] reported increased synthesis of IFN-α in children with Aicardi–Goutières syndrome (AGS), and defined the first Mendelian disease associated with enhanced type I IFN activation. In 2003, Crow et al. [11] assumed the existence of IFN-α hyperactivity as a common pathological keystone accounting for the overlapping features observed among apparently disparate human diseases, i.e. congenital HIV-1 infection, Mendelian encephalopathy AGS and autoimmune systemic lupus erythematosus (SLE). Growing insights were also derived from investigating the AGS molecular foundation [12], dissecting the type I IFN upregulation in monogenic forms of SLE and exploiting nucleic acid metabolism-driven IFN induction. In 2011, Crow et al. [1] described this new group of Mendelian disorders, termed interferonopathies, and defined them as novel inborn errors of immunity characterised by enhanced type I IFN signalling as a pathogenic keyplayer rather than a simple biomarker. Since then, the knowledge of the molecular mechanisms underlying these entities has widened exponentially leading to a “position framework” for type I IFN-mediated monogenic inflammation [2], representing the counterpart of other genetically determined immune signalling disorders, such as primary immunodeficiency [13] and monogenic auto-inflammation [14].
In the past few years, the list of putative monogenic interferonopathies has broadened as new molecular mechanisms and associated clinical phenotypes have been described [8, 15]. Type I IFN-driven inflammation may target a wide range of tissues and organs, including the lungs. A predominant auto-inflammatory or auto-immune phenotype may occur, respectively, according to the underlying signalling mainly employed (i.e. IFN-driven engagement of innate rather than adaptive immune responses) [15]. In the light of this concept, type I interferonopathies have been proposed to be part of the continuum model of self-directed immune disorders in which diseases rely on an auto-inflammatory-auto-immune spectrum. Nevertheless, despite growing evidence pointing to unleashed type I IFN signalling as the core of the so-called interferonopathies, a causal relationship to disease pathogenesis is still largely under debate [8].
Disease mechanisms of type I interferonopathies
The type I IFNs were originally described in 1957 as soluble molecules produced by cells in response to inactivated influenza virus exposure [16]. Once released, those cytokines were demonstrated to be capable of interfering with viral replication and infection onset. Since then, type I IFNs have been widely recognised as the first line of defence, empowering the host to counteract pathogen invasion by inducing a sustained antiviral inflammatory state and antiproliferative activity [5]. In order to achieve efficient immune defence that avoids exaggerated inflammation once the pathogen has been cleared, cells are equipped with timely switch-on/off intracellular machinery (figures 1 and 2). In the past few years, the identification of monogenic conditions displaying type I IFN upregulation and presenting with overlapping clinical pictures has provided invaluable insights on the multifaceted mechanisms that tightly regulate type I IFN production [3, 6, 8].

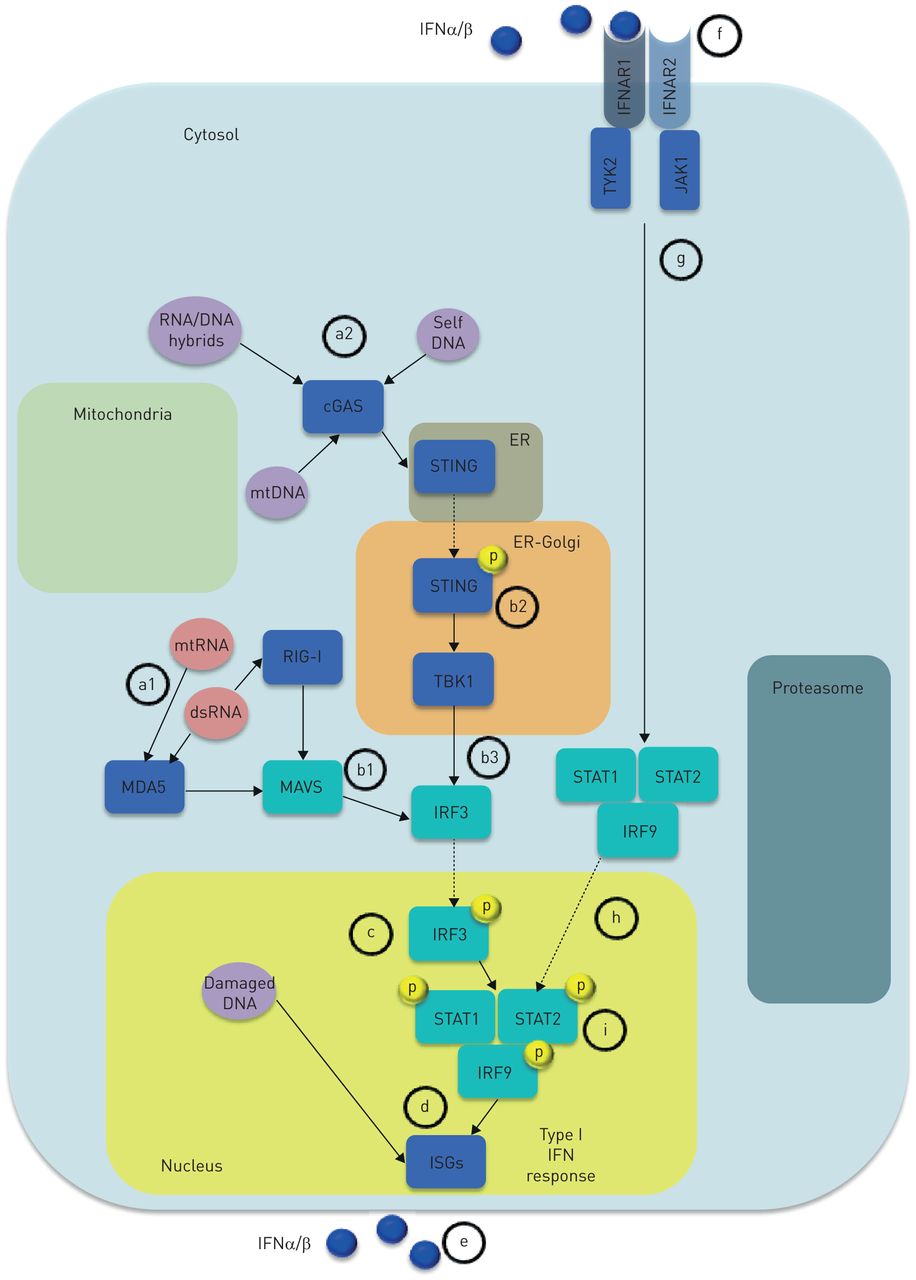
Type I interferon (IFN) response. Any nuclear cell is capable of producing type I IFNs in response to viral-derived or endogenous nucleic acids, sensed by distinct cytosolic PPRs, that signal through cytosolic adapters and downstream signals causing activation of IRFs. IRFs translocate to the nucleus and induce ISG expression resulting in type I IFN response and IFN production. a1) Detection of cytosolic dsRNA and mtRNA largely relies on members of the RLR family including MDA5 (also known as IFN-induced helicase C domain containing protein 1, IFIH1) and RIG-I. b1) MDA5 and RIG-I mediate RNA sensing through the cytosolic adapter MAVS/IPS1. Thus, upon engaging nucleic acids, RLRs undergo conformational changes thereby allowing interaction with their adaptor proteins leading to activation of IRFs that c) translocate to the nucleus thus inducing d) IFNαβ transcription. Less defined is the DNA sensing machinery. a2) Cytoplasmic dsDNA seems to interact with cGAS, leading to the production of cGAMP, which eventually engages the adapter molecule STING (also known as TMEM173) STING, located in the ER. b2) Once activated, STING translocates to the ER–Golgi compartment, where the signal is propagated through the phosphorylation of the TBK1. b3) Finally, TBK1 leads to c) IRF-3 activation that d) induces type I IFN response following IRF-3 nuclear translocation. Once released, e) type I IFNs (blue circles) may act via autocrine/paracrine manner f) by binding a single heterodimeric type I IFN transmembrane receptor composed of the subunits IFNAR1 and IFNAR2. Upon engaging one of the IFNR subunits, type I IFNs cause dimerisation of IFNAR1 and IFNAR2, leading to g) activation of the JAKs: TYK2 and JAK-1. Activated TYK2 and JAK1 h) drive the phosphorylation and subsequent translocation to the cell nucleus of the STAT family members (i.e. STAT1 and STAT2) and IRF9, resulting in formation of STAT1-STAT2-IRF9 ternary complex ISGF3. The final outcome is type I IFN response refuelling, consisting of ISG expression and IFN production. cGAS: cyclic GMP-AMP synthase; dsDNA: double-stranded DNA; ER: endothelial reticulum; IFNAR1: IFNα receptor-1; IFNAR2: IFNα receptor-2; ISG: IFN stimulated gene; IPS1: interferon-β promoter stimulator 1; IRF: IFN regulatory factor; JAK-1: Janus kinase-1; JAK: Janus kinase; MAVS: mitochondrial antiviral signalling; MDA5: melanoma differentiation-associated protein 5; mtDNA: mitochondrial DNA; mtRNA: mitochondrial RNA; PPR: pattern recognition receptor; RIG-I: retinoic acid-inducible gene I; RLR: RIG-I-like receptors; STAT: signal trasducer activator of transcription; STING: stimulator of interferon genes; TBK1, TANK-binding kinase 1; TYK2: tyrosine-protein kinase 2.
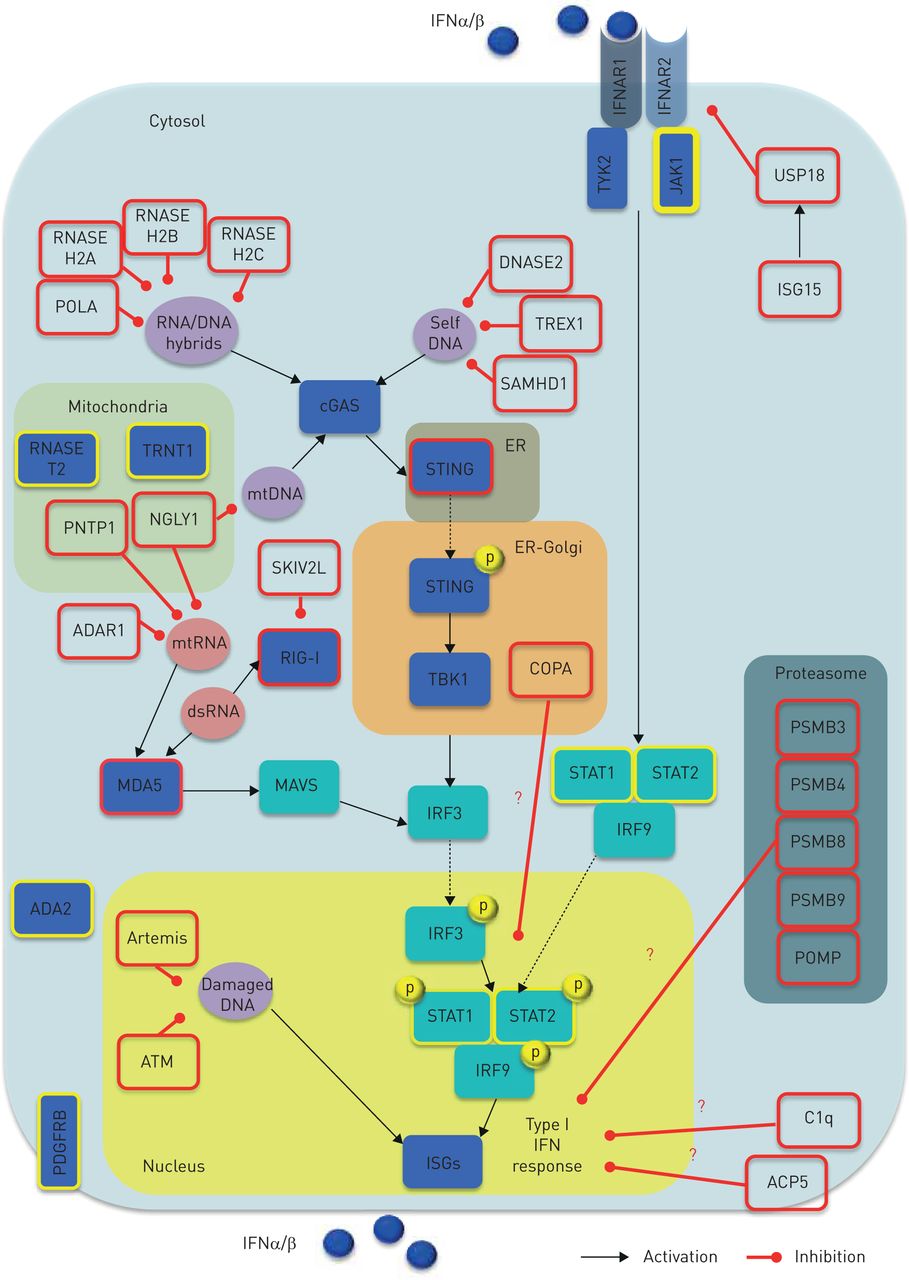
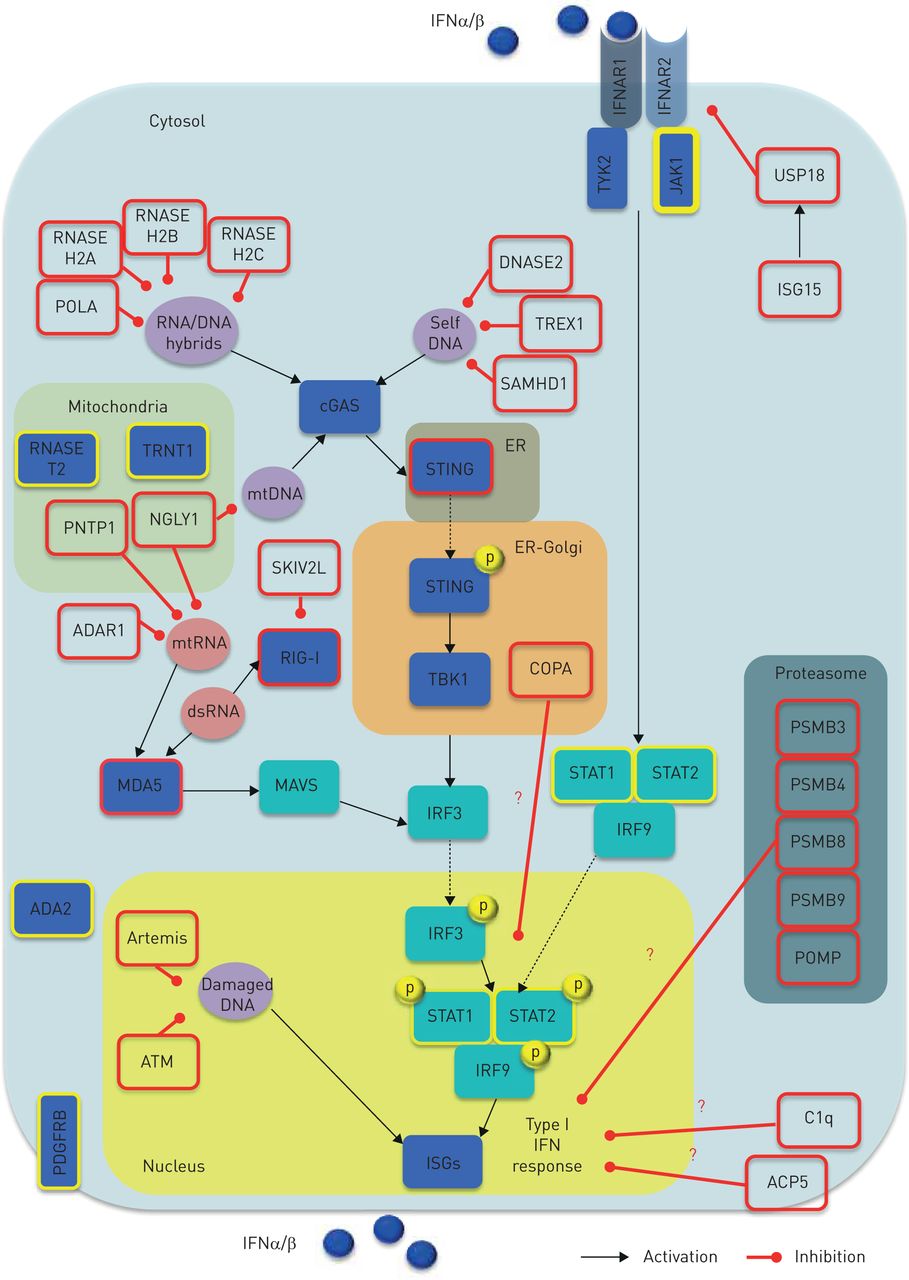
Type I interferon (IFN) activity regulation and monogenic interferonopathies. Both inappropriate activation (e.g. triggered by self-nucleic acids) and impaired negative regulation of the type I IFN system can give rise to interferonopathies (red border boxes). Nucleic acid-driven inflammation may originate either from disruption in the sensing machinery or PPR downstream mediators, as well as impairment of their processing, metabolism (including autophagy) and repair. In turn, aberrant activity of deoxyribonuclease, ribonuclease, DNA polymerase, RNA helicase, RNA editing machinery, polynucleotide phosphorylase, N-deglycosylation, dsDNA break repairs and ER-localised UPR may account for overproduction of type I IFNs. Furthermore, aberrancies in the proteasome component have been proposed to induce type I IFN signalling through indirect effects upon nucleic acid species processing. Overall, aberrant type I IFN enhancement may be due to either: 1) abnormal stimulation (e.g. increased accumulation or change in composition of endogenous nucleic acids); 2) aberrant sensing (e.g. constitutive activation or enhanced sensitivity of PPRs, leading to a change in threshold at which endogenous nucleic acids are sensed); 3) perpetuated activation (e.g. increased sensitivity or constitutive activation of IFN-inducing mediators other than PPRs); or 4) impaired negative regulation (e.g. unrestrained signalling due to defective negative feedback). A key negative regulator of this loop is represented by the ISG15, which causes induction of USP18 that, in turn, inhibits IFNAR2 activity, thus providing negative feedback to restrain an appropriate type I IFN pathway activity. Genes causing monogenic interferonopathies are depicted by red border boxes. Red question marks indicate unknown mechanisms. Yellow border boxes indicate genes suggested to cause type I IFN signalling aberrancies, but putative mechanisms are provisional. Black arrows indicate activation; red lines address inhibitory activity. ACP5: acid phosphatase 5; ADA2: adenosine deaminase 2; ADAR1: adenosine deaminase, RNA-specific, 1; ATM: ataxia-telangiectasia mutated gene; C1q: complement component C1Q; cGAS: cyclic GMP-AMP synthase; COPA: coatomer protein complex, subunit-α; dsRNA: double-stranded RNA; ER: endothelial reticulum; IFNAR1: IFNα receptor-1; IFNAR2: IFNα receptor-2; ISG: IFN stimulated gene; IRF: IFN regulatory factor; JAK-1: Janus kinase-1; JAKs: Janus kinases; MAVS: mitochondrial antiviral signalling; MDA5: melanoma differentiation-associated protein 5; mtRNA: mitochondrial RNA; NGLY1: N-glycanase 1; PDGFRB: platelet-derived growth factor receptor β; PNTP1: polyribonucleotide nucleotidyltransferase 1; POLA: POLA DNA polymerase-α; POMP: proteasome maturation protein; PSMB: proteasome subunit β; RIG-I: retinoic acid-inducible gene I; SKIV2L: superkiller viralicidic activity 2-like alias SK12 like RNA helicase; STAT: signal trasducer activator of transcription; STING: stimulator of interferon genes; TBK1, TANK-binding kinase 1; TRNT1: tRNA nucleotidyl transferase 1; TYK2: tyrosine-protein kinase 2; UPR: unfolded protein response; USP18: ubiquitin-specific peptidase 18.
Although a prompt stimulation of type I IFN is crucial to overcome viral infection, either inappropriate activation (e.g. triggered by self-nucleic acids) or impaired negative regulation of the type I IFN system may give rise to interferonopathies (figure 2). The current putative disease model of such entities is based on type I IFN pathway aberrant activation in response to either: 1) an excessive burden of nucleic acids derived from endogenous retro-elements; or 2) constitutive induction of nucleic acid sensors/mediators, leading to a nucleic acid-drive inflammation. Of note, the burden of nucleic acids may originate from either disruption in the sensing machinery or pattern recognition receptor (PRR) downstream mediators, as well as impairment of their processing, metabolism (including autophagy) and repair. Furthermore, aberrancies in the proteasome components have been proposed to induce type I IFN signalling through an indirect effect upon nucleic acid species processing (figure 2).
It is conceivable that for disruption in any driving step of the type I IFN system, homeostasis may account for type I IFN overproduction (figure 2). Whether or not the observed type I IFN enhanced signalling is actually the driving player or is an epiphenomenon is still highly debated [8]. In the past few years, several inborn monogenic errors affecting key components of the type I IFN machinery have been reported [17–92]. These clinical entities relate to a breakdown of self/nonself discrimination, involving mutant genotypes targeting molecules playing direct or indirect roles in nucleic acid signalling and IFN-driven inflammation. However, despite compelling data linking enhanced type I IFN signalling to clinical phenotypes, definitive evidence supporting the causal relationships to pathogenesis is still lacking [8]. Nevertheless, any diseases consistently associated with increased type I IFN activity underline the biological link between the observed disorder and type I IFN homeostasis disruption. The clinical spectrum of these entities is extremely heterogeneous. Furthermore, the number of clinical pictures is increasing within the context of known genotypes. Here, we provide an overview of the currently known main patterns of presentation featuring monogenic interferonopathies (supplementary table 1).
Known pulmonary features of recognised monogenic interferonopathies
Lung involvement in monogenic interferonopathies: patterns of presentation
In the last few years, several clinical reports have identified the lung as one of the key target organs of IFN-mediated inflammation. Pulmonary involvement may occur as a major clinical feature overlapping different genotypes associated with type I IFN-signalling disruption. Respiratory symptoms may be either mild or absent at disease onset and variably associated with systemic or organ specific inflammation (e.g. skin, central nervous system, vascular and gastrointestinal systems). While signs of ongoing systemic inflammation are usually present, associated extrapulmonary clinical features may precede lung function impairment by years. Thus, patients may display functional, radiological or histological signs of severe/endstage lung involvement, although these may be clinically hidden during long-term disease course. Conversely, a few cases of monogenic interferonopathies with atypical onset characterised by major lung involvement have been reported without clinically manifested extrapulmonary signs. To date, two major clinical patterns of lung involvement have been described during monogenic interferonopathies: 1) interstitial lung disease (ILD), including diffuse alveolar haemorrhage (DAH); and 2) pulmonary arterial hypertension (PAH) (table 1 and figure 3). However, advances in type I IFN signalling and its impact on targeted organs indicate other potentially related clinical pictures (e.g. bronchiectasis in STAT1), but further investigation is needed to confirm the actual clinical significance.

{kind=link}
{kind=link}
{kind=link}
Patterns of lung involvement in monogenic interferonopathies. interstitial lung disease (ILD), diffuse alveolar haemorrhage (DAH) and pulmonary arterial hypertension (PAH) as major patterns of lung manifestation in monogenic interferonopathies. SAVI, COPA, Aicardi–Goutières syndrome, DNAse II deficiency and CANDLE/PRASS are depicted by specific coloured boxes, listing putative genes so far identified in patients displaying severe pulmonary manifestation during monogenic interferonopathies. CANDLE: chronic atypical dermatosis with lipodystrophy and elevated temperatures; COPA: coatomer protein complex, subunit-α; PRASS: proteasome-associated auto-inflammatory syndrome; SAVI: STING-associated vasculopathy with onset in infancy; STING: stimulator of interferon genes.
ILD
ILD is recognised as the major pulmonary phenotype occurring during monogenic interferonopathy. To date, the ILD spectrum has been variably reported in STING (stimulator of interferon genes)-associated vasculopathy with onset in infancy (SAVI), chronic atypical dermatosis with lipodystrophy and elevated temperatures (CANDLE) and coatomer protein complex, subunit-α (COPA) syndrome (table 1).
ILD in SAVI
SAVI is an auto-inflammatory disease caused by a mutation in TMEM173 (encoding STING protein) and clinically characterised by systemic and peripheral vessel inflammation leading to distal tissue damage and cutaneous vasculopathy [15, 32, 93–101, 118–120] (table 1). In addition to persistent elevation of inflammatory markers, laboratory findings often include antinuclear antibodies (ANA) positivity and raised IgG and IgA. Since its initial description, pulmonary involvement has been a major clinical feature of SAVI [32]. All six patients described presented in early infancy with systemic inflammation and violaceous lesions of fingers, toes, cheeks and ears, eventually leading to acral necrosis and telangiectasia in most of the patients. Two patients presented with isolated tachypnea in the first weeks of life, and later developed typical cutaneous and systemic inflammation. Lung biopsies unveiled scattered mixed lymphocytic inflammatory infiltrate, interstitial fibrosis and emphysematous changes. Overall, five out of six SAVI patients displayed evidence of ILD, but two of them had no history of respiratory symptoms. Death from pulmonary complications and secondary infection occurred in other two patients. Autopsy performed in one of them demonstrated widespread vasculopathy of systemic and pulmonary vasculature [32].
There is variable clinical expression even in the presence of the same genotype [15, 62, 93–101, 118–120]. To date, total 30 patients with inherited TMEM173 gain-of-function mutations in variable ethnic backgrounds (e.g. mixed European, Japanese/northern European, Algerian, Caribbean, Hazaras, Turkish) have been described in 12 published works consisting of single case reports or small cohort studies [15, 93–101, 118–120].
Age of disease onset varies from neonatal to infancy. However, some patients with onset in adulthood have been reported. Despite signs of systemic inflammation and failure to thrive being essentially constant, associated clinical symptoms at onset ranged from typical cutaneous vasculopathy to less common clinical pictures such as lupus-like features with arthritis [93], symptoms mimicking childhood granulomatosis with polyangiitis [93], erythematosus-infiltrated skin lesions with pustular evolution followed by scarring, chilblains, and severe nail dystrophy [105] or isolated tachypnea [32, 95]. Mild respiratory symptoms (e.g. tachypnea, chronic cough, moderate distress associated with intermittent fever, recurrent bronchospasm, digital clubbing) often arose in the first months of life or preschool age, but were underestimated for a long time. Conversely, a few cases (two familial and one sporadic) of atypical onset characterised by severe pulmonary fibrosis as the first major manifestation have been reported without clinically manifested extrapulmonary signs [96].
Additional rare associated manifestations occurring during disease history included granulomatous hepatitis [98] and mild renal involvement (i.e. microscopic haematuria and mild proteinuria) with hypertension [101] reported in two patients. In two cases, given the history of intrauterine growth retardation, failure to thrive, early onset of recurrent bacterial respiratory infections, and severe septicaemia [98] or early-onset recurrent low-grade fever, dermatitis and diarrhoea [101], immunodeficiency was initially suspected and ruled out. Given the rarity of the disorder and the extreme variability of its onset and disease history, diagnosis was always reached after years/decades since the clinical onset, often facilitated by the occurrence of typical onset and definitive diagnosis in a family member or, sometime, post mortem pathology findings. When assessed (nine out of 28), the blood IFN signature (i.e. evaluation of increased expression of IFN stimulated genes (ISGs whose transcription is induced by IFN) was consistently positive, suggesting type I IFN pathway activation [93, 96, 100, 101]. Variants in pulmonary fibrosis causing genes (i.e. SFTPC, SFTPB, ABCA3, TERT, TERC and NKX-2) were analysed in two familial cases [96] and turned out negative.
Because of the variable presentation, pulmonary function tests (PFTs) were not routinely performed. However, data available from seven patients demonstrated a predominant restrictive or mixed pattern with reduced diffusing capacity for carbon monoxide (DLCO) [96, 100, 101]. When obtained, bronchoalveolar lavage fluid demonstrated inflammatory infiltrate with large amount of lymphocytes or pattern consistent with neutrophilic alveolitis [93]. Given the occurrence of pulmonary manifestations during the disease course, most of the patients eventually underwent chest computed tomography (CT). Data available from eight patients [93, 96–98] variably demonstrated diffuse ground glass and reticular opacity [93, 98], honeycombing/cysts with ground glass areas [96], hilar and paratracheal lymphadenopathy, lung fibrosis and emphysema [96], ground glass with lung fibrosis [97], focal thickening of the interlobular septa with areas of ground-glass opacities with predominant subpleural distribution [101].
Overall, 24 out of 30 SAVI patients had disease courses characterised by ILD, in one case associated with secondary PAH [94]. Thus, lung biopsy was eventually performed in the majority of the patients, and variably showed: macrophage alveolitis, follicular hyperplasia and interstitial fibrosis [118]; neutrophils and macrophages within chronic alveolar and interstitial inflammation with following progressive fibrosis and associated PAH [94]; active inflammation with type 2 pneumocyte hyperplasia and lymphocyte infiltrate [95]; multiple pulmonary nodules in central alveolar peribronchiolar area, including lymphocytic inflammatory infiltrate forming aggregates with associated fibrosis without vasculitis [96]; bronchiectasis associated with lymphoid infiltrate and interstitial fibrosis [96], additional examination with electron microscopy performed in one case [95] unveiled endothelial tubureticular inclusions, which were suggestive for type 1 IFN excess. Sudden worsening of pulmonary functions with manifested evidence of ILD and endstage pulmonary fibrosis was frequently described while ILD-associated PAH was reported in one case [66]. All the patients had disease activity poorly controlled by ongoing immunosuppressant therapies, and in a few cases, lung disease required patients to be listed for early lung transplant [32, 96]. Regardless the ongoing therapeutic strategies, death for pulmonary complications with secondary infection or respiratory insufficiency, and multi-organ failure following lung transplant have been described [32, 96, 99].
ILD in CANDLE syndrome
CANDLE syndrome is an auto-inflammatory disorder caused by autosomal recessive loss-of-function mutations in the proteasome subunit beta type 8 (PSMB8) [15, 66–68]. Following its initial description, it has become evident that loss-of-function PSMB8 variants may account for several distinct conditions (table 1), and similar clinical disorders have been related to mutation of other components of the proteasome (i.e. PSMB9, PSMB4, PSMA3, POMP) [15, 69–74]. Hence, CANDLE syndrome is now considered to be part of the wider spectrum defined proteasome-associated auto-inflammatory syndrome (PRASS) [65, 79]. Usually, CANDLE/PRASS syndrome clinical features variably include recurrent fever, severe growth retardation, violaceous periorbital changes, panniculitis-induced lipodystrophy, mild lymphocytic meningitis, headache, basal ganglia calcifications, ILD, nonerosive synovitis, arthralgia, myositis, recurrent infections, cytopenias, systemic hypertension, dyslipidaemia, elevated acute phase reactants and hypergammaglobulinaemia (table 1). CANDLE syndrome was first described in four Spanish/Hispanic patients [66]. One of the four subjects displayed ILD but further clinical information on pulmonary pattern is lacking. This patient was treated with a variety of anti-inflammatory and immunosuppressive therapies but the disease progressed with sudden death at 14 years of age. Cause of death was unclear. The patient's family history was unremarkable until the age of 12 years, when her newborn sister manifested similar cutaneous manifestation without lung involvement. Subsequently, the five CANDLE syndrome patients clinically reported [66, 67] were genetically screened [68]. PSMB8 pathogenic variants were then identified in seven out of eight subjects with clinical presentations consistent with CANDLE syndrome and disease onset by 6 months of age. Beside the patient already described, a 5 year-old American/Caucasian with CANDLE syndrome also displayed lung involvement, consisting of an organising pneumonia (OP)-like pattern. In a review paper [15], an additional CANDLE syndrome patient previously unpublished and affected by interstitial pneumonitis was also reported. Overall, ILD may rarely occur in CANDLE/PRASS [15, 79]. However, subclinical pulmonary involvement was not routinely investigated, and thus cannot be definitely excluded [15, 65–82].
ILD and alveolar haemorrhages in COPA syndrome
ILD represents a major clinical manifestation in COPA syndrome. COPA syndrome is due to mutations in the COPA gene, which encodes the α-subunit of the coatomer protein complex (COPI), involved in transiting molecular cargo from the Golgi to the endoplasmic reticulum [61, 105]. Despite the fact that the encoded protein is ubiquitously expressed, this syndrome typically targets joints, lungs and, to a lesser extent, the kidneys [61, 105–115, 121]. All patients present with early onset of inflammatory arthritis of small and large joints, variably associated with ILD and pulmonary haemorrhages. A subset of patients have been reported to have immune-mediated kidney disease (table 1). High-titre autoantibodies (i.e. antimyeloperoxidase and ANA) are usually present as well as increased Th17 cells and interleukin (IL)-1β/ IL-6 expression. Originally reported in 21 genetically confirmed patients [61], average age of presentation was 3.5 years (6 months–22 years) and 16 subjects had disease onset before the age of 5 years. All patients had lung disease diagnosed as pulmonary haemorrhage, ILD or both, and lung pathology findings consistent with pattern observed in systemic autoimmune syndromes. The potential role of IFN in COPA syndrome pathogenesis was first highlighted by Volpi et al. [107] who demonstrated a positive IFN signature in five genetically confirmed patients, opening novel therapeutic opportunities. As in other interferonopathies, the diagnosis is often challenging. As described in the following reports, clinical picture mimicking juvenile idiopathic arthritis (JIA) may occur as isolated early manifestations [108, 109]. Onset of pulmonary symptoms and progressive lung damage may appear only years [108, 109] or decades [109] after disease presentation, often facilitated by familial cases with similar symptoms [109]. The clinical picture is poorly responsive to standard immunosuppressive therapies, leaving lung transplant often as the only therapeutic option. Conversely, initial presentation may resemble idiopathic pulmonary hemosiderosis with delayed definitive diagnosis only at the late appearance of morning stiffness and nonerosive arthritis [110].
Although recognised as a main clinical feature of the syndrome, the pulmonary features of COPA syndrome patients have not been comprehensively described until lately, when Tsui et al. [111] systematically analysed a cohort of 14 subjects, with onset within 12 years of age,i.e. <1 year (7%), 2–9 years (71%), 10–12 years (21%). Initial presentation ranges from isolated joint pain (three out of 14), pulmonary symptoms (six out of 14) or a combination of the two (five out of 14). Four out of 14 had haemoptysis as initial lung manifestation while two patients presented with anaemia and fatigue, due to DAH. Those without haemoptysis presented with chronic cough, shortness of breath and dyspnoea on effort. Eventually, all subjects developed ILD and arthritis without uveitis. Among those who had isolated articular onset, all developed lung symptoms within 10–20 years. Thus, subjects were given several clinical diagnoses prior to discovery of COPA syndrome including JIA, rheumatoid arthritis and idiopathic pulmonary haemosiderosis. Seven out of 14 patients developed DAH. Notably, a biopsy confirmed immune-mediated kidney disease and renal failure was present in three out of seven subjects with DAH. PFTs performed in all the patients unveiled a prevalent restrictive pattern (eight out of 14) but obstructive (one out of 14) and mixed (two out of 14) profiles were found in a minority, with normal lung function in one patient. Initial DLCO % predicted was abnormal for all subjects tested (eight out of eight). Longitudinal PTF surveillance was available for 10 subjects, and showed decline in predicted FEV1 and forced vital capacity (FVC) over time for nearly all subjects (nine out of 10), with average of 2.6% and 1.8% per year for FEV1 and FVC, respectively. Only one of the 14 patients did not undergo CT imaging since this patient had early onset suggestive of ILD diagnosed via lung biopsy. Overall, CT findings included cysts (described as thin-walled and scattered throughout the parenchyma in a variable distribution) (nine out of 11), ground-glass opacities (six out of 11), nodules (five out of 11) and fibrosis (one out of 11). CT scans showed stabilisation or amelioration while on various immunosuppressive regimens but radiological improvement did not correlate with lung function recovery. Histopathological features on lung biopsy were follicular bronchiolitis (seven out of 10), DAH (four out of 10), acute interstitial fibrosis in a non-usual interstitial pneumonitis pattern (two out of 10) and airspace enlargement/cystic changes (two out of 10). All seven with follicular bronchiolitis had ground glass opacities and/or nodules on CT scan. Three subjects did not receive any lung biopsy while three were biopsied at the time they developed haemoptysis and diagnosed with DAH via bronchoscopy. Two of these subjects had evidence of acute lung injury with capillaritis. Interestingly, earlier age of onset was observed in successive generations in a three-generation kindred later described [98]. Presenting symptoms were cough and dyspnoea in four out of four, and histopathological findings included small lung cysts, follicular bronchiolitis, ILD and neuroendocrine cell hyperplasia. Neither alveolar haemorrhage nor glomerular disease were present. Features not previously associated with COPA syndrome included neuromyelitis optica, pulmonary carcinoid tumour, clear cell renal carcinoma, renal cysts, hepatic cysts, nephrolithiasis, pyelonephritis and meningitis. Kidney involvement may not be present [121]. Conversely, lupus nephritis was the clinical manifestation at onset in a 10-year-old girl with unremarkable medical history [113]. Her disease course included transient arthralgia without joint/synovial lesions, and no signs of early lung involvement. COPA syndrome was suspected based on positive history of glomerulonephritis, arthritis and lung haemorrhages variably occurring in her family members, eventually leading to genetically confirmed COPA syndrome diagnosis. She developed endstage renal failure while on immunosuppressive therapies.
PAH
PAH occurrence has been described in both ab estrinseco and constitutive exposure to type I IFNs, highlighting the potential role of IFNα/β in PAH pathogenesis. PAH has been associated with increased IFN signature in its early phases of development, and it can be induced by therapeutic use of type I IFNs in several disorders [122]. In addition, PAH has been described in monogenic interferonopathies including SAVI, CANDLE syndrome, AGS and DNaseII deficiency (figure 3), as well as in multifactorial diseases associated with increased IFN signalling, such as SLE. Although the disease model and causal link of IFN-induced PAH is still to be defined, ex vivo treatment with a selective JAK2 inhibitor reduces proliferation of human arterial endothelial cells in idiopathic PAH [122]. Furthermore, potential efficacy of the JAK1/2 inhibitor ruxolitinib has been recently described in IFN-associated PAH in children with SAVI or DNaseII deficiency. Even if the underlying mechanism is still largely undefined, PAH represents one of the major pulmonary complications (although rare) observed in some monogenic interferonopathies.
PAH in SAVI
PAH was firstly reported in a male patient of Japanese/northern European ancestry with SAVI [94]. Following severe systemic inflammatory onset during the first weeks of life, the child developed the typical vasculitic rash associated with persistent elevation of inflammatory markers. Lung biopsy performed at age of 2 years displayed neutrophils and macrophages within chronic alveolar and interstitial inflammation. The child lately developed progressive pulmonary fibrosis and associated PAH. High-dose corticosteroid treatment provided partial beneficial effect, mainly on respiratory symptoms. Disease-modifying and immunosuppressive agents as well as biologic drugs were tried but failed to provide clinical improvement, and the child died of respiratory failure at the age of 16 years. PAH was later described in another two SAVI patients who were of Chinese/Malasyan ancestry. Disease onset and course were variable. While one patient history was characterised by early-onset growth retardation, chilblain lesions on the ear, skin telangiectasia and long, clubbed fingers, associated with chronic dry cough along with progressive decreased activity tolerance leading to ILD and associated PAH in adolescence [102], the other patient displayed a more abrupt onset with life-threatening PAH at 3 years of age [103]. The child had previously suffered of acute respiratory distress at the age of 2 months during interstitial pneumonitis due to Pneumocystis jirovecii, followed by full recovery. He later presented failure to thrive, developmental delay, livedo reticularis and vesicular rash without cutaneous vasculitis nor increased inflammatory markers. Echocardiogram performed during follow-up at 21 months of age was normal. At 33 months, the child developed a PAH and was started on a pulmonary vasodilator (i.e. sildenafil). Given the unusual presentation, the IFN gene signature was assessed and found positive, and genetic testing identified a de novo missense TMEM173 variant. Interestingly, in this case the presence of a concomitant ILD could not be assessed, and whether or not the observed PAH was isolated or secondary to IFN-related lung fibrosis is still an open question.
PAH in CANDLE syndrome
The first case of non-ILD-related PAH in these disorders was described in a 2-year-old Guatemalan female from a consanguineous union, who was eventually diagnosed with CANDLE syndrome [104]. The child presented with failure to thrive, facial lipodystrophy, recurrent fevers associated with steroid-dependent neutrophilic dermatosis lesions. A chest radiograph highlighted cardiomegaly and an echocardiogram demonstrated PAH. Chest CT scan ruled out a concomitant ILD but unveiled dilated pulmonary arteries consistent with the presence of PAH. In addition, acute thrombosis of the superior vena cava and left brachiocephalic vein was reported in the CT scan, although antiphospholipid antibodies were present at low titres. Given the lack of evidence of thromboembolic disease or the presence of autoantibodies, the degree to which the acute thrombosis contributed to the development of PAH remains undefined. Targeted sequencing of PSMB8 identified homozygous missense variant, which is a founder mutation in Hispanic patients.
PAH in Aicardi–Goutières syndrome
Although previously unrecognised, a recent retrospective analysis of an AGS cohort unveiled that PAH may occur also in this set of monogenic diseases [116]. AGS is an early onset, progressive encephalopathy characterised by basal ganglial calcifications, white matter abnormalities and chronic cerebrospinal fluid lymphocytosis (table 1). Interestingly, four out of 22 individuals with complete medical information were shown to have developed PAH. Three carried IFIH1 gain-of-function mutations while one had a heterozygous TREX1 mutation. Typical symptom onset was between birth and 10 months of age at the latest, and PAH presentation variably occurred between the first month and 16 years. All the patients displayed concomitant elevated IFN signature scores, with the highest levels in those carrying IFIH1 variants. Thus, genetic variants of IFIH1 and TREX1 have been suggested to be putative causes of congenital PAH.
PAH in DNaseII deficiency
Recently, onset of PAH at 17 years of age was described in a patient affected by DNaseII deficiency, and this occurrence appeared to be IFN-mediated [123]. DNaseII deficiency is characterised by a spectrum of clinical features including neonatal anaemia, membranoproliferative glomerulonephritis, liver fibrosis, deforming arthropathy and increased anti-DNA antibodies [34] (table 1). The patient had a history of neonatal hepatopathy, cytopenia, lupus pernio, growth retardation, recurrent fever, polyarticular arthritis, chronic glomerulonephritis and lipodystrophy. At 14-years-old, the patient's IFN signature was suggestive of an underlying interferonopathy, and a pathogenic DNaseII mutation identified by whole exome sequencing (WES). The patient's clinical picture progressively declined in spite of several therapeutic strategies. The introduction of off-label treatment with ruxolitinib allowed a dramatic clinical improvement and steroid tapering. However, after 5 months of treatment, and about 1 month following a likely viral illness, the patient was diagnosed with PAH, raising the question of whether this was drug-related or due to IFN-related inflammation during an uncontrolled disease activity. In fact, at the time of PAH diagnosis, a dramatic increased of IFN-score had been observed. Furthermore, a combination therapy with steroid pulses, epoprostenol, sildenafil and furosemide failed to ameliorate the clinical picture. Conversely, once ruxolitinib was reintroduced at higher doses a dramatic improvement of pulmonary pressure was achieved and the patient's cardiologic follow-up showed slow, progressive further recovery.
Diagnostic approach
As evident by the clinical overview on monogenic interferonopathies described here, diagnosis can be extremely challenging due to the high variability in disease onset and course. In addition, diagnostic tools for definitive diagnoses are not available worldwide, and validation of most of them is still ongoing, mainly in research hospitals. Since severe lung impairment may occur clinically hidden during monogenic interferonopathies, useful hints may be derived from a detailed medical history, focusing on the occurrence of early-onset systemic inflammation variably associated to a wide spectrum (supplementary table 1) of extrapulmonary manifestations (e.g. suggestive cutaneous manifestations, peripheral vasculitis, panniculitis, nasal septum perforation, failure to thrive, recurrent fevers, suggestive neurological involvement, arthritis, contractures, autoimmune kidney disease, cytopenias, recurrent infections). In addition, family history of ILD, autoimmune kidney disease or arthritis as well as suggestive cutaneous manifestations in a young patient with early-onset systemic inflammation, should drive attention to potential underlying inborn errors of the IFN pathway. Conversely, early onset of either respiratory symptoms with suggestive family history or suggestive tout court lung involvement (i.e. ILD, alveolar haemorrhages, PAH or bronchiectasis) should prompt a hypothesis-driven work-up aimed at assessing the presence of monogenic interferonopathy (supplementary figure 1). Once clinically suspected, enhanced type I IFN signalling should be tested for. However, the development of diagnostic tools to reach a definitive diagnosis of type I IFN-related disease has been hampered by the current inability to directly detect and quantify type I IFN protein in biological samples using the available ELISA assays. Recently, an ultrasensitive single molecule array for detecting human IFNα in biological fluids has been developed and validated [117]. However, to date, the most employed available tool is the so-called IFN signature [124, 125], which assesses the increased expression of ISGs whose transcription is induced by IFN. It consists of a 6- or 28-IFN response gene scoring, based on QRT-PCR or NanoString technology. Of note, the IFN signature is not specific to type I IFN. The possibility that enhanced IFN signalling induced by types II and III IFNs might also have detrimental effects deserves further investigation. Despite its limitations, including interlaboratory variability, the IFN signature may help to screen patients for IFN-aberrancies, thus identifying those worth of genetic testing [124–126]. Recently, a clinical score correlating with the ISG score has been developed, providing a potential additional tool to guide in decision making for diagnostic work-up in monogenic interferonopathies [127]. In the presence of suggestive clinical picture, disease course and family history, especially if an IFN-positive signature has been identified, panel next-generation sequencing or targeted Sanger sequencing should be performed to make a definitive diagnosis, based on the known potential genes involved (table 1, figures 2 and 3). Furthermore, given the fact that some intereferonopathies may have severe lung impairment as the presenting clinical picture, a young patient presenting with early-onset lung involvement such as ILD, alveolar haemorrhages or PAH, should be investigated for IFN-related genes as well as those already known for congenital lung diseases (figure 3), even in the absence of suggestive extrapulmonary manifestations (supplementary figure 1).
Therapeutic options
Given the recognised key role of nucleid acid-induced type I IFN response and inflammation, current therapeutic approaches mostly rely on strategies aiming at: 1) restraining the production of endogenous nucleic acids (i.e. reverse-transcriptase inhibitors (RTIs)); 2) limiting their sensing (i.e. cGAS inhibition by hydroxychloroquine [128]); 3) promoting their clearance (mTOR inhibitors [129]); or 4) inhibiting downstream signalling (i.e. TBK1 inhibition [130, 131] anti-type I IFN antibodies [132], JAK inhibitors [55, 97, 100, 101, 102–104, 119, 123, 133, 134]).
A recent clinical trial of RTIs in AGS patients provided promising results, indicating an effect of restraining IFN signalling [135]. To date, the most clinically explored approach in a wide spectrum of interferonopathies is that described with JAK1/2 inhibition (i.e. tofacitinib, ruxolitinib, baricitinib), which displayed promising beneficial effects on lung manifestation in patients with SAVI, COPA syndrome, CANDLE syndrome and DNaseII deficiency [55, 97, 100, 101, 102–104, 119, 123, 133, 134, 136, 138]) (table 2).
Effects of interferonopathy (IFN)-signalling blockade on lung involvement in monogenic interferonopathies
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary_table_1.pdf ERR-0001-2020_Supplementary_table_1
Footnotes
Number 3 in the Series “Rare genetic interstitial lung diseases” Edited by Bruno Crestani and Raphaël Borie
This article has supplementary material available from err.ersjournals.com
Previous articles in the Series: No. 1: Daccord C, Good J-M, Morren M-A, et al. Brit–Hogg–Dubé syndrome. Eur Respir Rev 2020; 29: 200042. No. 2: Hadchouel A, Drummond D, Abou Taam R, et al. Alveolar proteinosis of genetic origins. Eur Respir Rev 2020; 29: 200187.
Provenance: Commissioned article, peer reviewed.
Conflict of interest: S. Cazzato has nothing to disclose.
Conflict of interest: A. Omenetti has nothing to disclose.
Conflict of interest: C. Ravaglia has nothing to disclose.
Conflict of interest: V. Poletti has nothing to disclose.
- Received January 2, 2020.
- Accepted May 27, 2020.
- Copyright ©ERS 2020.
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References









