





Abstract
Multiple synchronous lung nodules are frequently encountered on computed tomography (CT) scanning of the chest and are most commonly either non-neoplastic or metastases from a known primary malignancy. The finding may initiate a search for primary malignancy elsewhere in the body. An exception to this rule, however, is a class of rare primary lung neoplasms that originate from epithelial (pneumocytes and neuroendocrine), mesenchymal (vascular and meningothelial) and lymphoid tissues of the lung. While these rare neoplasms also present as multiple synchronous unilateral or bilateral lung nodules on chest CT, they are often overlooked in favour of more common causes of multiple lung nodules. The correct diagnosis may be suggested by a multidisciplinary team and established on biopsy, performed either as part of routine diagnostic work-up or staging for malignancy. In this review, we discuss clinical presentations, imaging features, pathology findings and subsequent management of these rare primary neoplasms of the lung.
Abstract
Several rare primary lung neoplasms may present as multiple synchronous indeterminate lung nodules on chest CT. These merit increased awareness among radiologists, pathologists and pulmonologists, as well as a multidisciplinary team approach. http://bit.ly/2QpRH1D
Introduction
Multiple synchronous incidental unilateral or bilateral lung nodules are frequently encountered on routine chest computed tomography (CT) scans. Lung cancer screening with low-dose CT scans has further increased the detection of such nodules. Morphology, size and distribution of nodules are features that are often helpful in differentiating benign from neoplastic aetiologies. In the majority of cases, multiple lung nodules are grouped into two main categories: non-neoplastic and metastatic neoplasms from a known or an unknown primary malignancy. However, there is a third category of rare primary lung neoplasms that also present as multiple synchronous pulmonary nodules. These lesions originate from the normal cells within the lung that undergo clonal transformation at multiple foci throughout the lung, thus presenting as multiple pulmonary nodules. This review highlights the clinical, imaging and morphological features of these lesser-known neoplastic, non-metastatic causes of multiple lung nodules detected on chest CT. Our review aims to encompass the diagnosis and management of these multiple rare synchronous lung cancers. Since benign conditions, such as infection or sarcoidosis, and metastatic cancers to the lung are more common causes of multiple lung nodules seen on CT, this uncommon group of synchronous lung neoplasms can be exceedingly challenging to diagnose. The rarity of these diseases and lack of medical literature to guide treatment contribute to the lack of awareness among physicians. We rationalise that this review will increase awareness of this group of rare entities, encourage the need for discussion in a multidisciplinary setting, help expedite the diagnostic process and prompt appropriate management. We have intentionally excluded deposition diseases such as amyloidosis, which can also manifest as multiple bilateral lung nodules but are considered non-neoplastic. A classification system based on the cell of origin of these rare tumours is depicted in table 1.
Classification system based on the cells of origin of multiple synchronous lung nodules
Lymphoproliferative disorders
Primary pulmonary lymphomas are rare neoplasms compromising <1% of all non-Hodgkin lymphomas and 0.5–1% of all primary pulmonary malignancies. They include pulmonary extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT) lymphomatoid granulomatosis, post-transplant lymphoproliferative disorder (PTLD) and diffuse large B-cell lymphoma (DLBCL) [1].
Pulmonary extranodal marginal zone lymphoma of MALT
MALT lymphoma, which makes up approximately 70–90% of the primary pulmonary lymphomas, is considered an indolent neoplasm with a potential for slow growth and dissemination over a prolonged period of time. It has a slight female predominance [2, 3] and a favourable prognosis with a 2-year survival of 100% and a 5-year survival close to 80% [4–11]. The median (range) age at the time of diagnosis is 61 (range: 21–80) years, and over half of patients are smokers. Many patients present with an asymptomatic mass on imaging studies. When present, symptoms include cough, minor dyspnoea, night sweats, weight loss, chest pain and, rarely, haemoptysis [12].
Pulmonary MALT lymphomas have an association with smoking [13] and disease states characterised by chronic lung inflammation, such as hypersensitivity pneumonitis, diffuse panbronchiolitis and rheumatoid arthritis [14–17], supporting a theory of antigenic stimulation resulting in bronchial lymphoid tissue proliferation.
Multiple unilateral or bilateral pulmonary nodules, lung masses and/or airspace consolidation are the most common CT imaging features in up to 70% of patients (figure. 1). There may be associated peribronchial thickening and air bronchograms with bronchial distension. Lymphangitic spread into the surrounding parenchyma and hilum, as well as mediastinal lymphadenopathy, are other imaging features that may be seen. Rarely, cystic lesions or pleural effusions are reported [6, 7, 18].
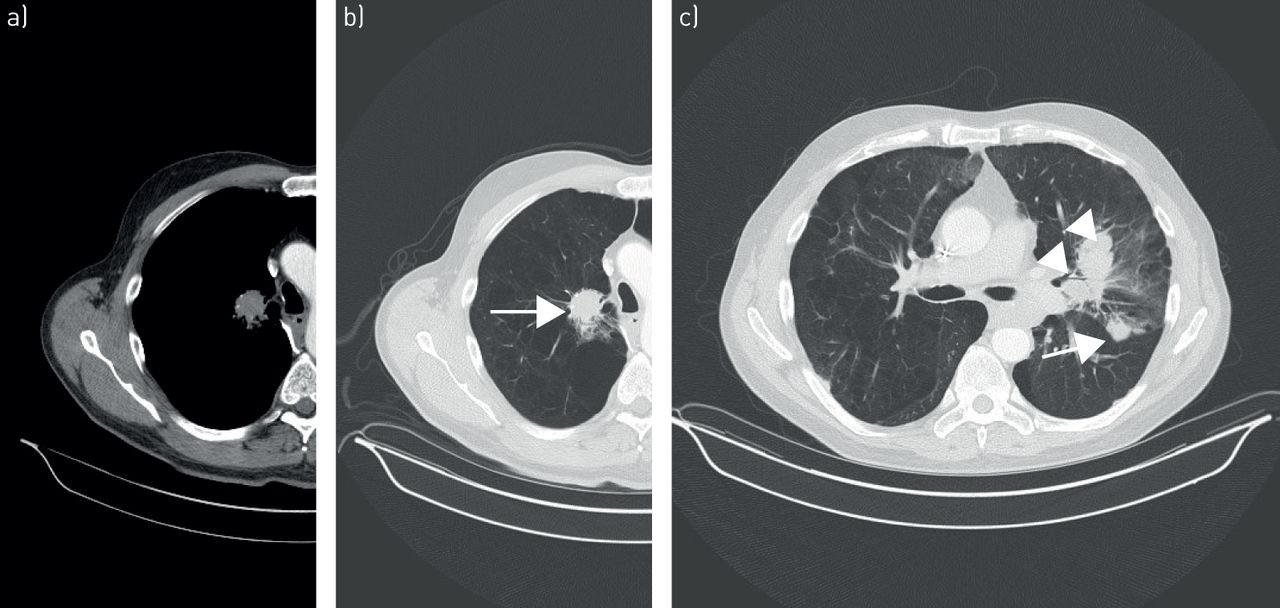
a–c) Pulmonary extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue. Contrast-enhanced computed tomography scan in lung and mediastinal window settings reveal multiple bilateral solid nodules (arrows) and mass-like consolidations (arrowheads) with irregular margins and surrounding ground-glass opacities, suggestive of lymphangitic spread in a, b) the right paramediastinal and c) left perihilar region.
The diagnosis is made by histological examination of specimens obtained from surgical biopsies – either thoracotomy or video-assisted thoracoscopy. Transbronchial biopsies have a significantly lower yield. Morphological features include the presence of lymphocytes with proliferation around the bronchovascular interstitium and subpleural areas (figure 2a). Invasion into the bronchioles, vessels and pleura is a common finding that distinguishes MALT lymphomas from exuberant reactive infiltrates (figure 2b). The infiltrate is comprised of diffuse, small lymphocytes, plasmacytoid lymphocytes, so-called centrocyte-like B-cells and monocytoid B-cells (figure 2c). These cells may overrun adjacent reactive follicles, so-called follicular colonisation. Immunohistochemical staining reveals a predominance of CD20+ lymphocytes (B-cells), as well as diffuse positivity for CD13, CD79a and BCL2. The infiltrates are negative for CD5 and CD10 staining [6, 7, 19]. The neoplasm can be associated with the presence of MALT-lymphoma-specific genetic aberration, the t(11;18)(q21;q21) resulting in API2/MALT1 fusion transcripts [11, 20].
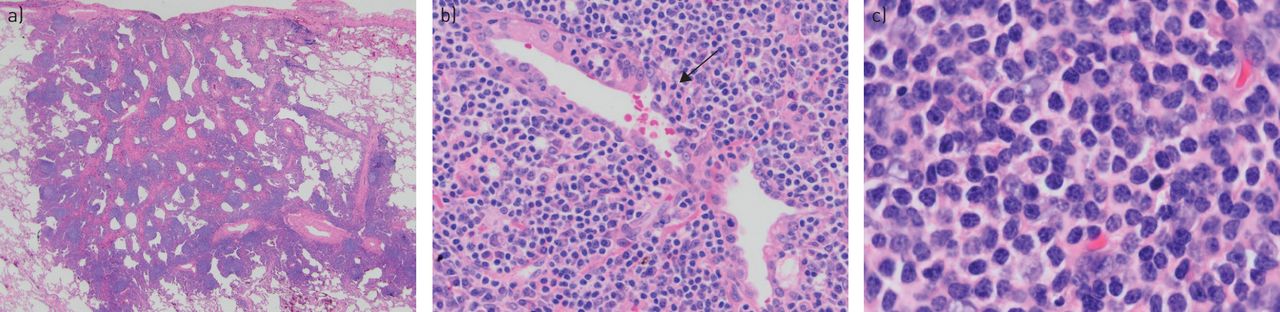
Pulmonary extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue. a) Diffuse infiltration of a lymphocytic infiltrate of small lymphocytes with scattered follicles (haematoxylin and eosin; 12.5×). b) The lymphoma invades adjacent structures, including airways (arrow), a feature that distinguishes this infiltrate from a benign reactive inflammatory infiltrate (haematoxylin and eosin; 100×). c) The malignant cell population consists of small lymphocytes with clear spaces between adjacent cells, so-called centrocytic-like features. Larger nuclei and plasmacytoid cells may also be seen (haematoxylin and eosin; 400×).
Given the indolent course of this tumour, immediate treatment following diagnosis may not be indicated. TROCH et al. [11] reported a retrospective study involving 11 patients, all of whom remained untreated after a median follow-up period of 28.1 months. Eventually, three of those patients showed progression and were placed on therapy. Moreover, this untreated group was compared with 10 patients on treatment during the same period. These 10 patients underwent various treatment modalities, and after a median follow-up time of 58 months, one patient from the treated group died. No significant difference was seen in terms of time to progression [11]. AHMED et al. [6] studied 22 patients, 20 of whom were treated. Treatment modalities reported were surgery, chemotherapy with or without rituximab and radiation therapy. The chemotherapy regimens used were CHOP (cyclophosphamide, Adriamycin, vincristine and prednisone), RCD (rituximab, cyclophosphamide and dexamethasone), CVP (cyclophosphamide, vincristine and prednisone) and RF (rituximab and fludarabine). Of those 20 patients, 19 achieved an objective response described as a complete resolution in nine patients and 10 achieving a partial response, while one did not respond [6]. However, the literature shows no clear data on the clinical course of MALT lymphoma when treated versus left untreated, likely related to this tumour being very rare.
Lymphomatoid granulomatosis
Lymphomatoid granulomatosis is a rare Epstein–Barr virus (EBV)-associated B-cell lymphoproliferative disorder characterised histologically by the angiocentric and angiodestructive proliferation of atypical lymphoid cells. It tends to spread along the bronchovascular bundles and interlobular septa, often resulting in distortion of the underlying lung architecture. First described by LIEBOW et al. [21] in 1972, these lesions are composed of EBV-positive B-cells surrounded by a reactive T-cell infiltrate [22, 23].
Most patients are middle-aged men with a male to female ratio of 2:1 [21, 24]. Symptomatic patients reported having cough, often productive, dyspnoea and fever. Other symptoms include weight loss, arthralgia and skin lesions. Skin involvement can be in the form of an erythematous macular rash, small plaque-like lesions or indurated subcutaneous lesions predominantly on the extremities [21]. Clinically, the lungs are involved in 100% of the patients, most commonly in the lower lobes, followed by the central nervous system (38%), liver (19%), skin (17%) and kidneys (15%). There is usually no nodal, bone marrow or spleen involvement [24].
The most commonly reported radiographic finding is bilateral nodular opacities in the mid-lower lung zones. The nodules often show angiocentricity and peribronchovascular distribution (figure. 3). Most can progress rapidly, coalesce and may cavitate. Migratory nodules may be seen in some instances, with some of these nodules exhibiting a spontaneous “waxing–waning” course. Other findings include unilateral nodular opacities, ill-defined pulmonary opacities, air bronchograms, thin-walled cysts and hilar lymphadenopathy [18, 21, 25–29].

Lymphomatoid granulomatosis. Computed tomography scans a) at and b) below the carinal bifurcation level demonstrates multiple bilateral angiocentric and peribronchovascular lung nodules.
LEE et al. [29] reviewed imaging studies of five patients with lymphomatoid granulomatosis. The main chest radiograph finding was nodules or masses with poorly defined margins. The number of nodules ranged from one to 19 and size from 1–5 cm. CT chest scans showed bilateral nodules ranging in number from five to >60 with irregular yet well-defined margins. Most nodules were <1 cm, and the largest diameter ranged from 1.3 to 6.5 cm. In one case, magnetic resonance imaging revealed masses with higher intensity signals than muscles on the T1- and T2-weighted images. In addition, a right upper lobe pulmonary artery narrowing caused by a perihilar mass and severe left main pulmonary artery wall thickening resulting in complete occlusion by either thrombosis or the tumour was seen.
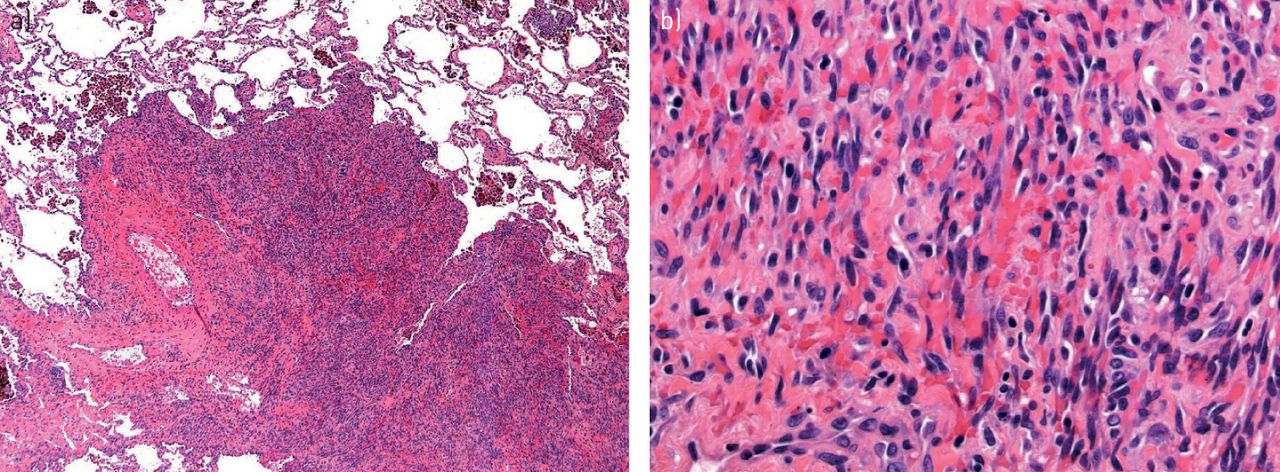
Given the nonspecific radiological findings, histology is needed to confirm the diagnosis. Findings include the presence of angiocentric immunoproliferation of well-demarcated, mixed mononuclear cells that have a predilection to infiltrate the lumen and the subintimal regions of medium-sized arteries and veins (figure 4) [21, 25, 29]. Immunohistochemical and in situ hybridisation techniques are used to identify EBV-infected cells, as most cases of lymphomatoid granulomatosis have shown proliferation of EBV-infected large B-cells with a prominent T-cell reaction. The presence of these findings are key features necessary for the diagnosis of lymphomatoid granulomatosis [22, 23, 30, 31]. Lymphomatoid granulomatosis is graded based on the number of EBV-positive large B-cells per high power field: grade 1: <5 positive cells; grade 2: 5–50 cells; and grade 3: >50 positive cells [32, 33].

Lymphomatoid granulomatosis. a) Large, subpleural nodule with prominent necrosis (arrow) (haematoxylin and eosin; 12.5×). b) Pleomorphic lymphoid population invades pulmonary artery (arrow) and adjacent airway (haematoxylin and eosin; 40×).
Treatment of lymphomatoid granulomatosis is guided by the grading, so it is imperative to have an accurate evaluation and diagnosis of the specimens. Interferon-α is used for grade 1 and 2 disease, whereas immunochemotherapy with dose-adjusted etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, rituximab is used for grade 3 disease. The National Cancer Institute performed a prospective trial studying the outcomes in patients following treatment with interferon-α based on the grading. Progression-free survival improved to 56% (median follow-up of 5.1 years) in patients with lymphomatoid granulomatosis grade 1 and 2 and 44% (median follow-up of 32 months) in patients with grade 3 disease [34].
PTLD
PTLD occasionally develops in recipients of solid organs and rarely among bone marrow recipients. It usually appears within a year of transplant (1 month to 7 years). The clinical course varies from a benign flu-like illness to potentially life-threatening fulminant disease and mirror pathological features, ranging from benign lymphoid hyperplasia to frank malignant lymphoma. Risk factors for development include EBV seronegativity, concomitant cytomegalovirus infection, allograft type, paediatric age and immunosuppressive regimen. Polyclonal B-cell lymphomas (positive EBV population cell lines) are more common than monoclonal lymphomas in these specimens [35–37]. Extranodal involvement is more common than nodal involvement, and imaging features depend on the organ of involvement. Lung involvement usually manifests as multiple homogeneous solid nodules of varying sizes (figure 5), which may occasionally cavitate. Diffuse involvement of the lungs, pleural thickening and mediastinal lymphadenopathy are other features that may be seen on CT [18, 38].

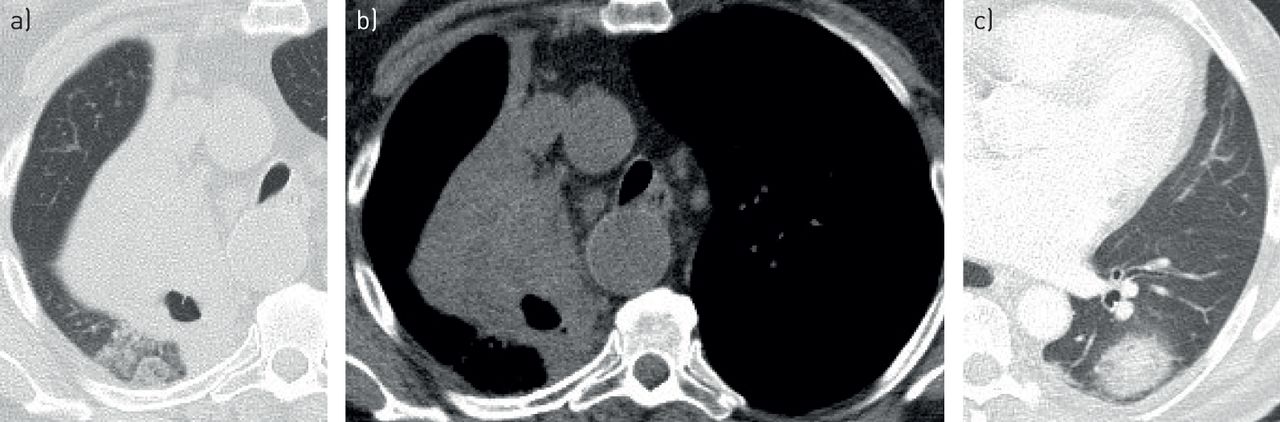
Post-transplant lymphoproliferative disorder. Computed tomography images shows multiple randomly distributed solid nodules in all lung lobes of a young male patient with renal transplant in the past 12 months.
In the lung, PTLD is usually divided into two categories: polymorphic (low-grade) or monomorphic (high-grade) lesions. The gross morphology of these nodules usually reveals tan/white nodules with necrosis. On histopathology, large lymphoid nodules with pleural invasion may be seen (figure 6a). Polymorphic PTLD consists of small lymphocytes, immunoblasts and plasma cells with low-grade cytological features, which are distinguished from reactive infiltrates by the presence of EBV-positive cells. Monomorphic PTLD consists of lymphoid infiltrates with predominantly large cells with areas of marked cytological atypia and mitoses. Invasion of surrounding structures may be seen (figure 6b) [39, 40].

Post-transplant lymphoproliferative disorder (PTLD), high-grade (monomorphic) type. a) Large, subpleural lymphoid nodule with invasion into the overlying pleura (haematoxylin and eosin; 12.5×). b) Lymphoid infiltrate has large cells with areas of marked cytological atypia and mitoses, consistent with a high-grade PTLD (haematoxylin and eosin; 200×).
Reduction of immunosuppression is the cornerstone of therapy. Disease regression in response to decreasing immunosuppressive regimen is a diagnostic feature. Other treatment modalities include surgical resection of local disease, radiotherapy and medical treatment with interferon-α, antiviral therapy, interleukin-2 infusion and rituximab. The prognosis depends on the pathological grade and cell type with the low-grade polymorphic type behaving more favourably than high-grade PTLD in the lung [41].
DLBCL
DLBCLs that are primary to the lung are unusual. They comprise approximately 10–20% of all pulmonary lymphomas and are seen in patients aged 50–70 years with no sex predisposition [42]. A number of risk factors have been identified, including collagen vascular diseases, immunosuppression, congenital immunodeficiencies and AIDS. Patients usually present with cough, haemoptysis and dyspnoea and may report fever and cachexia.
Imaging studies reveal single or multiple solid nodules or a mass, which commonly cavitates or shows central hypoattenuation (figure 7). Thoracic lymphadenopathy may be present. Tissue diagnosis is needed to confirm the diagnosis. Small biopsies, including cytological specimens, can be diagnostic as the cells have large, malignant vesicular nuclei and high-grade cytological features (figure 8). The cells express a B-cell phenotype (CD20 and CD79a) with scattered reactive T-cells in the background. The morphological features can be similar to carcinoma, melanoma and germ cell tumours. Therefore, immunohistochemical studies and flow cytometric analysis are essential to confirm the diagnosis. Distinguishing a primary pulmonary DLBCL from a mediastinal large B-cell lymphoma extending to the lung is difficult and requires both clinical and imaging findings. Also, since DLBCL may be EBV positive, it should be distinguished from lymphomatoid granulomatosis and the high-grade form of PTLD [42].
a–c) Diffuse large B-cell lymphoma. Computed tomography scans in two different patients. a, b) Solid mass with central areas of necrosis and cavitation in the right supra-hilar region resulting in right upper lobe atelectasis. c) Part-solid nodule in the left lower lobe with peripheral lymphangitic spread.

Diffuse large B-cell lymphoma. a) Lymphoid nodule with marked necrosis invading bronchovascular area (haematoxylin and eosin; 20×). b) Histological features indistinguishable from high-grade post-transplant lymphoproliferative disorder (haematoxylin and eosin; 200×).
Vascular tumours
Epithelioid haemangioendothelioma
Epithelioid haemangioendothelioma is a rare tumour of vascular origin. It was first described by DAIL et al. [43]. WELDON-LINNE [44] confirmed its endothelial lineage by using electron microscopy and immunohistochemical techniques to show diffuse cytoplasmic staining with factor VIII-related antigen. WEISE et al. [45] coined the term epithelioid haemangioendothelioma based on these findings. It is a low to intermediate grade neoplasm, which can arise in different organ systems, such as the lungs, liver, bone or lymphoid tissue, and sometimes as synchronous primary tumours. Over half of the patients have symptoms at their initial presentation that are usually related to the size and location of the tumour, including cough, pleuritic chest pain, haemoptysis and shortness of breath [46]. It is more common in females with a mean age of onset between 35 and 44 years, with the majority being diagnosed before the age of 40 years. Life expectancy ranges between 1 and 15 years [47–49]. In one study, the 5-year survival was reported to be 60% [48]. Poor prognostic factors include weight loss, anaemia, pleural haemorrhage and effusion, fibrinous pleuritis, extrapleural proliferation and the presence of spindle tumour cells [47, 48].
Radiographically, it can present as unilateral or bilateral sub-centimetre perivascular nodules, with ill-defined or well-defined margins. Some nodules can grow up to 2 cm in size and can be seen in conjunction with pleural effusions. They are mostly found in relation to small- to medium-sized vessels and bronchi [50]. These findings can lead to an erroneous diagnosis of metastatic disease from another primary site (figure 9). Ground-glass opacities (GGOs) and interlobular septal thickening are other atypical radiological findings [49–51].

a, b) Epithelioid haemangioendothelioma. Computed tomography lung images demonstrate a) innumerable bilateral perivascular nodules. b) Coronal reformatted soft-tissue images also show multiple hepatic low-density lesions with nodular peripheral enhancement suggesting haemangiomas (arrow).
The diagnosis is confirmed by tissue sampling via a transbronchial, transthoracic, thoracoscopic or an open lung biopsy. The pathological findings include round to oval shaped nodules with a hypocellular centre and peripheral zones with bland epithelioid cells. The cells may contain intracytoplasmic lumina and can have an extracellular matrix in the surrounding tissue (figure 10). Immunohistochemistry can be positive for multiple vascular-endothelial markers including ERG, CD31, CD34, factor VIII and FLI1 [52–54]. Cytokeratin can be positive in these cells. Recently, translocations involving chromosomal regions 1p36.3 and 3q25 resulting in the formation of a WWTR1-CAMTA1 fusion gene have been discovered in approximately 90% of these cases. A polyclonal antibody to the C-terminus of CAMTA1 has proven to be a useful tool in the diagnosis of this neoplasm [55].

Epithelioid haemangioendothelioma. a) Bland, epithelioid cells within an eosinophilic matrix and rare lumina in cytoplasm (arrow) (haematoxylin and eosin; 40×). b) Cells have epithelioid morphology and many have prominent intracytoplasmic vacuole consistent with lumina formation (arrow) (haematoxylin and eosin; 200×).
Epithelioid haemangioendothelioma can metastasise after resection, and there is no standardised therapy for epithelioid haemangioendothelioma [56]. Surgical resection for isolated lesions, chemotherapy with radiation, immunotherapy for diffusely disseminated metastatic disease and watchful waiting for asymptomatic stable disease are all suggested treatment modalities [52, 57–61].
Kaposi sarcoma
Primary Kaposi sarcoma of the lung is extremely rare. Metastasis to the lungs from mucocutaneous or nodal disease is more common. There are four variants, of which the AIDS-related form with CD4 <200 cells·mm−3 is most commonly encountered. Human herpes virus (HHV)-8, as well as HIV, are presumed causative factors. The disease can also manifest in post-transplant immunosuppressed patients [62, 63].
Imaging findings include multiple, bilateral and fairly symmetrical, ill-defined, peribronchovascular lung nodules, “flame-shaped” opacities on high-resolution CT with perihilar and lower zone predominance, GGOs, patchy consolidations and interlobular septal thickening (figure 11). Pleural effusion and thoracic lymphadenopathy might also be present [64, 65]. Violaceous lesions can be found in the central airways on bronchoscopic examination [66].
Kaposi's sarcoma. Computed tomography images shows characteristic ill-defined peribronchovascular small nodules, patchy peribronchial consolidations (flame shaped), interlobular septal thickening and mild ground-glass opacities in both lungs with mid and lower zone predominance.
The pathological diagnosis can not only be made by thoracoscopic biopsies, but also by bronchoscopic and transthoracic needle biopsies. The diagnostic features of the lesion include atypical spindle cells with only minimal cytological atypia, intervening “slit-like” vascular spaces containing red blood cells in a “box-car” configuration and scattered plasma cells (figure 12). Immunohistochemical studies reveal the spindle cells are positive for HHV-8 and vascular antibodies including ERG and CD31 [67].

Kaposi's sarcoma. a) Spindle cell proliferation surrounding bronchovascular area with areas of haemosiderin pigment present in the peripheral lung (haematoxylin and eosin; 20×). b) Atypical spindle cells line vascular space with red blood cells (haematoxylin and eosin; 200×).
Angiosarcoma
Pulmonary angiosarcoma is a rare tumour and is largely metastatic. Thorium dioxide (Thorotrast), used as a radiocontrast agent in the 1930s and 1940s and since discontinued, was heavily linked to hepatic angiosarcomas. Polyvinyl chloride is also a risk factor. Primary pulmonary angiosarcomas are considered exceedingly rare, and only a handful of cases have been reported. The most common imaging presentation is multiple lung nodules or masses associated with perilesional alveolar haemorrhage, pleural effusions and disseminated disease (figure 13) [68, 69].

Angiosarcoma. Contrast-enhanced computed tomography scan, axial and coronal reformatted images in a 74-year-old male demonstrate multiple, randomly distributed bilateral lung nodules of varying sizes with ground-glass halo (arrow), felt to represent perilesional alveolar haemorrhage. Interestingly, most nodules demonstrated central vascular enhancement on mediastinal windows (not shown).
Angiosarcomas throughout the body can have spindled and/or epithelioid morphology [70]. The pathological features of angiosarcomas in the lung have a more epithelioid morphology with cells more in clusters and solid nests. The nuclei are usually more vesicular, mimicking carcinomas and mesotheliomas (figure 14). Immunohistochemical studies reveal these epithelioid tumour cells can be cytokeratin positive, making the distinction from nonsmall cell carcinomas difficult. In this scenario, the ample vascular spaces in the background should raise the differential diagnosis of a vascular tumour and immunohistochemical studies for vascular markers (ERG and CD31) will be positive in angiosarcomas and negative in carcinomas.

Angiosarcoma. Malignant epithelioid cells with scattered areas of vascular spaces (haematoxylin and eosin; 100×).
Neuroendocrine tumours
Diffuse idiopathic pulmonary neuroendocrine cell hyperplasia
Pulmonary neuroendocrine cells are present throughout the pulmonary tree. These cells can proliferate as an adaptive response to hypoxia such as in people living at high-altitudes and patients with underlying chronic lung disease such as emphysema or fibrosis [71]. The term diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH) is given when there is no known predisposing factor for their proliferation [72]. The proliferation of these cells can result in distortion of the lung microstructure, as these cells have the ability to synthesise and release various amines and peptides.
DIPNECH is a rare under-recognised entity. According to the World Health Organization (WHO) classification of lung tumours, it is classified as a pre-invasive lesion within the spectrum of the epithelial neuroendocrine tumours [73]. The term DIPNECH was first used in 1992 by AGUAYO et al. [72], who first described six patients with diffuse hyperplasia of pulmonary neuroendocrine cells. Since none of their cases had any previous lung disease or smoking history, these authors suggested that hyperplasia in these patients was a primary process and the term DIPNECH was used. The majority of patients were middle-aged nonsmoking females, who present in most instances with nonspecific respiratory symptoms that have been present for years prior to diagnosis. Davies et al. [74] reviewed 19 cases; 79% were females and 84% were nonsmokers, with a mean age of 57.5 years. NASSAR et al. [75] reported 25 cases of which 92% were females with a mean age of 58 years, 67% were nonsmokers and 92% were symptomatic (71% had cough, 63% dyspnoea and 25% presented with wheezing). The pulmonary function test analysis showed 54% to have an obstructive pattern, 13% restrictive, 17% mixed obstructive and restrictive, and 17% with a normal pulmonary function test [75]. Diffusion capacity has also been shown to be compromised [72].
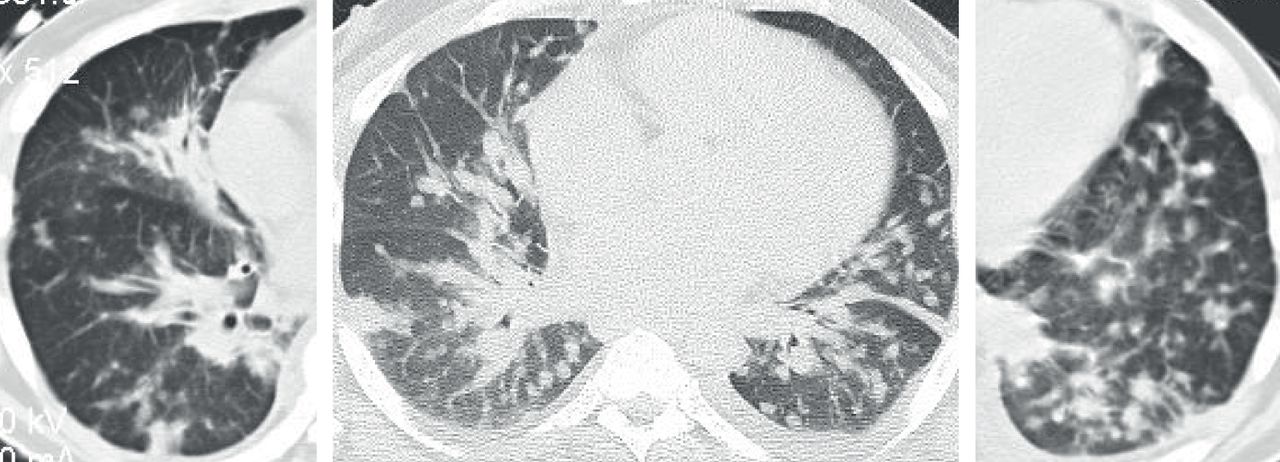
CT findings range from normal imaging to multiple bilateral pulmonary nodules (3 mm–3 cm), GGOs, bronchial wall thickening, bronchiectasis and mosaic attenuation with air-trapping [72, 74–76]. Presence of multiple small lung nodules with heterogeneous mosaicism of the lung parenchyma on routine inspiratory CT chest images and multifocal air-trapping on expiratory high-resolution CT may serve as diagnostic clues to the entity (figure 15).

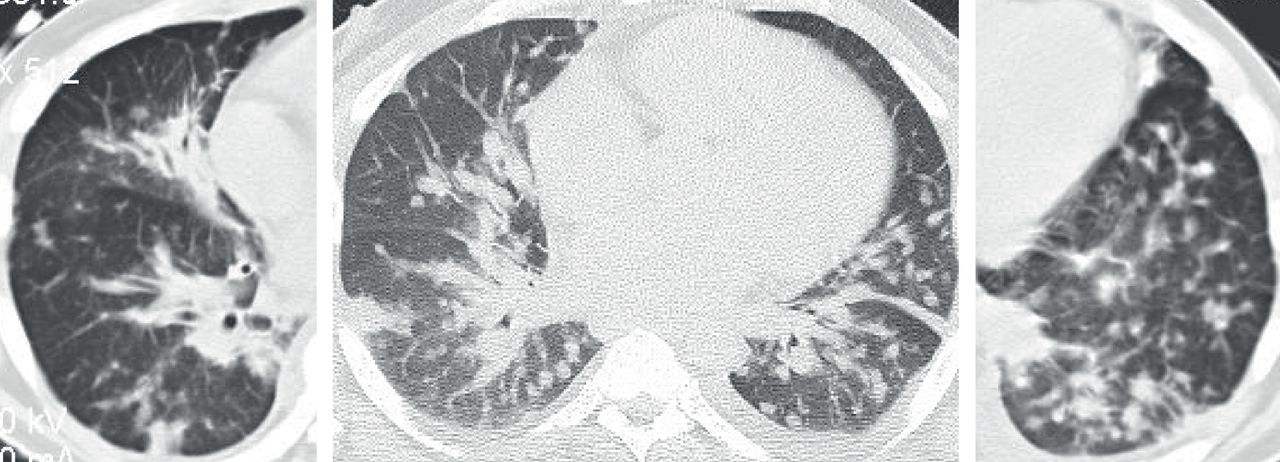
Diffuse idiopathic neuroendocrine cell hyperplasia. Axial computed tomography images demonstrate multiple ≤5 mm nodules (arrows) with subtle “mosaic pattern” of lung attenuation suggestive of air-trapping.
The diagnosis requires histopathological confirmation, and surgical biopsy is the gold standard. Pathological features include clusters of neuroendocrine cell hyperplasia within the epithelium of the small airways and tumourlets consisting of clusters of neuroendocrine cells <5 mm invading through the basement membrane into the subepithelial area. When these clusters are ≥5 mm, they are classified as a carcinoid tumour (figure 16). MARCHEVSKY et al. [77] proposed that “presence of five or more neuroendocrine cells, singly or in clusters located within the basement membrane of the bronchiolar epithelium of at least three bronchioles, combined with three or more carcinoid tumourlets can be used to consistently diagnose DIPNECH on a surgical biopsy”.

Diffuse idiopathic neuroendocrine cell hyperplasia. a) Neuroendocrine cell proliferation (arrow) present within a small airway with definitive extension beyond the basement membrane (haematoxylin and eosin; 20×). b) Immunohistochemical study of chromogranin highlights diffuse, strong staining of neuroendocrine cells (anti-chromogranin antibody; 40×). c) Aggregate of neuroendocrine cells forming 5-mm nodule, consistent with tumourlet formation (haematoxylin and eosin; 40×). d) Diffuse, strong staining of tumourlet for chromogranin (anti-chromogranin antibody; 40×).
To date, the treatment strategy is uncertain. Watchful waiting, chemotherapy, inhaled and systemic steroids, bronchodilators, lung resection and lung transplant have been used [72, 74, 75, 78, 79]. AGUAYO et al. [72] used chemotherapy in two patients, which showed no obvious clinical effect; one patient remained stable for 8 years and one died of respiratory failure.
The general course of the disease is stable, although clinical deterioration and progression to respiratory failure have been described. Thus, treatment should be tailored based on clinical presentation, course of the disease, presence of hypoxia, extent and distribution of the radiological findings and abnormalities of the pulmonary function test.
Meningothelial tumours
Minute pulmonary meningothelial-like nodules
This is a rare benign neoplasm of the lung of uncertain clinical significance. It never metastasises and can present as solitary or bilateral diffuse nodules. Previously known as chemodectoma because of characteristic microstructure, cytological features and presence adjacent to vessels, it is now known that these nodules contain endocrine granules and resemble meningothelial cells [80–83].
In most cases, the presentation is incidental, and patients are usually asymptomatic. The incidence of such tumours ranges between 0.3% and 9% of all lung resections [84–86]. The incidence was reported to be 7% in one of the largest reports studying 1724 pulmonary resection specimens. A total of 271 minute pulmonary meningothelial-like nodules were identified in 121 patients [86]. Approximately 80% of patients are females with most cases diagnosed in the 5th and 6th decades [83, 86, 87].
These tumours are found in association with lung injury, supporting a reactive rather than neoplastic origin. They are commonly found in patients with congestive heart failure, venous thromboembolism, chronic bronchitis, emphysema and lung cancer, particularly adenocarcinoma [83, 85, 86, 88].
On imaging, these tumours are mostly reported as scattered small nodules or micronodules (<2 mm in size) on CT (figure 17). They can present as solitary nodules or diffuse bilateral ground-glass nodules in a miliary pattern. On occasion, they can have an apical distribution [88]. In another report, it was found that these nodules tend to favour the right lung three times more than the left [83].

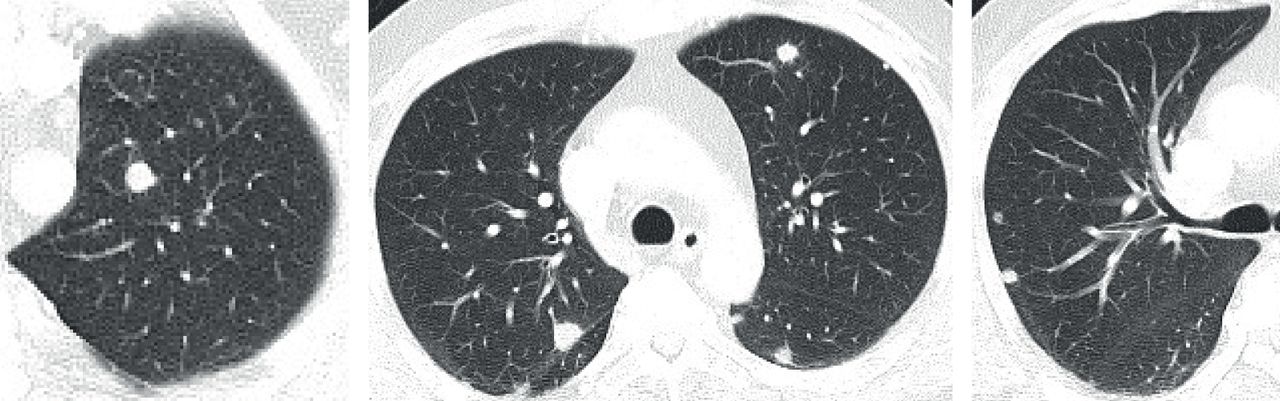
Minute pulmonary meningothelial-like nodules. Computed tomography images reveal multiple micronodules (<3 mm) in both lungs (arrows).
Microscopically, these nodules consist of epithelioid cell nests in a “Zellballen”-like arrangement and are located in relation to small veins [89]. The cells are round to oval and widen the alveolar wall (figure 18). On electron microscopy, these cells are connected with very well-formed desmosomes [83]. Immunohistochemical studies show positive staining for progesterone, vimentin and epithelial membrane antigen, which are markers found in meningothelial cells [84, 90].

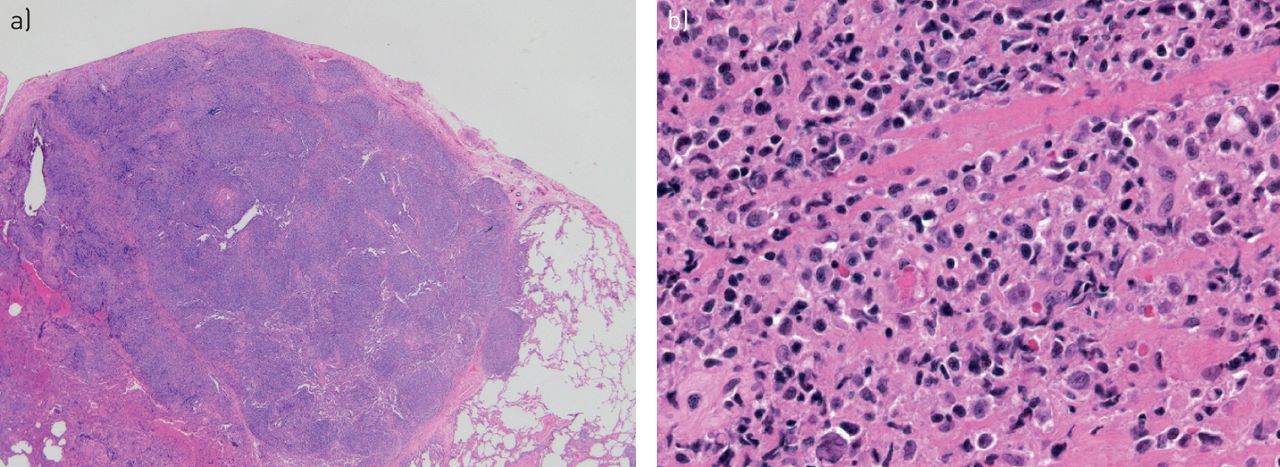
Minute pulmonary meningothelial-like nodules. a) Single lesion centred around a small venule in a thoracoscopic biopsy with multiple lesions (haematoxylin and eosin; 40×). b) Lesional cells have bland cytological features and a whorled “zellballen”-like architecture (haematoxylin and eosin; 200×).
The clinical significance of this rare, benign non-metastasising tumour is uncertain. Nonetheless, its association with other medical comorbidities should be highlighted and may warrant further workup.
Epithelial tumours
Adenocarcinoma in situ
Multifocal low-grade primary lung adenocarcinomas can present as multiple GGOs. The spectrum of noninvasive adenocarcinoma includes foci of atypical adenomatous hyperplasia (AAH) that measure ≤5 mm, adenocarcinoma in situ (AIS) if the focus measures >5 mm but ≤3 cm in greatest dimension, and minimally invasive adenocarcinoma (MIA) if the focus measures ≤3 cm and has an invasive or solid stromal component <5 mm. This represents a continuum of disease progression for all adenocarcinomas that evolve from GGOs (AAH, AIS and MIA) into solid nodules (invasive adenocarcinoma). Importantly, this progression may arise in multiple foci in the lung and, therefore, represent multiple primary tumours.
Adenocarcinoma in situ is a slow-growing, indolent subtype of lung adenocarcinoma. It is characterised by a lepidic, noninvasive, non-metastatic growth pattern. Previously referred to under the banner of “bronchioloalveolar cell carcinoma”, this term has now been discontinued, following the International Association for the Study of Lung Cancer/European Respiratory Society/American Thoracic Society recommendations from 2011 [91]. In the WHO classification of lung cancer from 2015, AIS has been categorised in the “preinvasive” category along with AAH [92].
High-resolution CT in AIS reveals pure GGOs <3 cm, usually ranging from 5 to 20 mm [92]. They have a higher attenuation and may be larger than GGOs seen with AAH, which are, by definition, <5 mm. An accurate assessment of these nodules requires thin-slice high-resolution CT with slice-thickness <3 mm (ideally 1–1.5 mm), to characterise ground-glass and part-solid components in the nodule [92]. They tend to be solitary and may show only minimal changes on radiographic follow-up. As such, they need long-term follow-up when compared to solid nodules in the lung if surgical resection is deferred (figure 19).
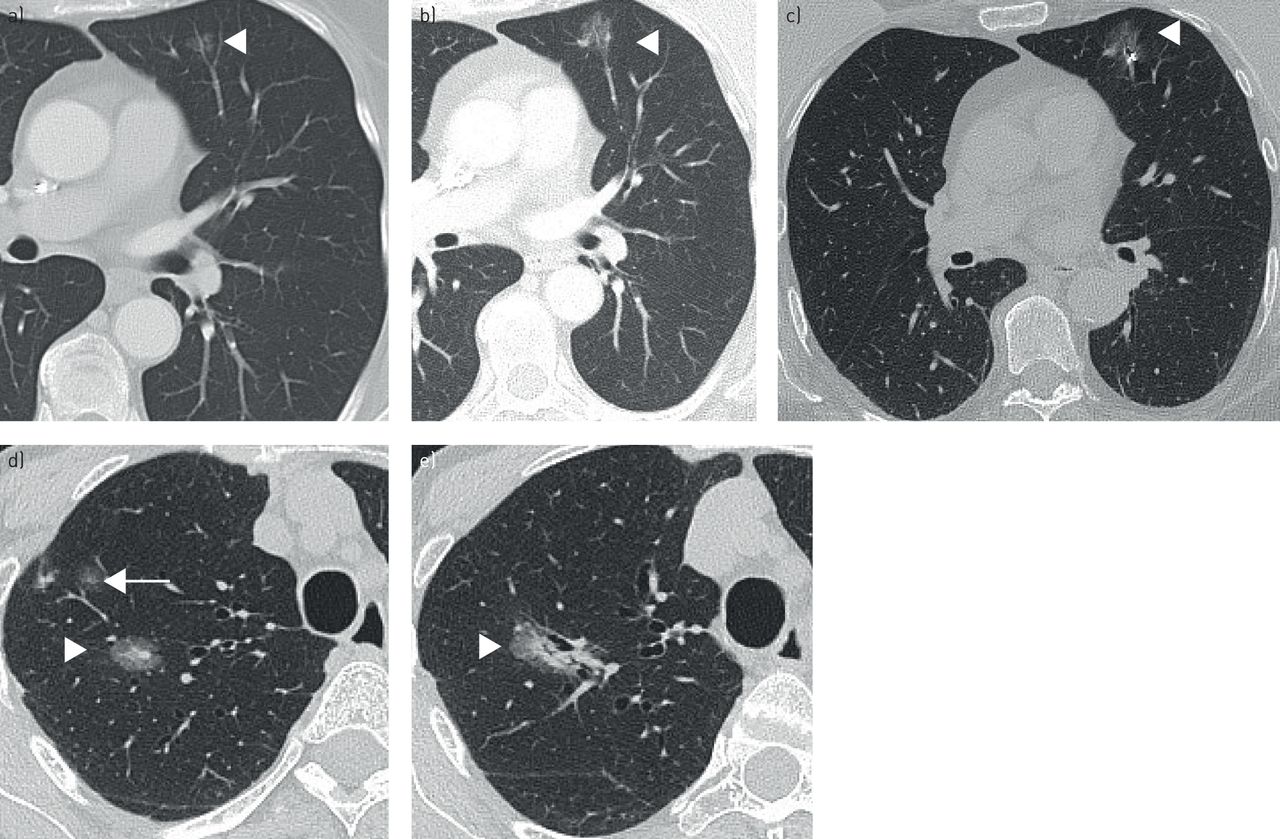
Spectrum of lung adenocarcinoma. a–c) Low-grade, indolent adenocarcinoma. a) Initial high-resolution computed tomography (HRCT) image reveals a 5-mm ground-glass nodule in the left upper lobe that could represent atypical adenomatous hyperplasia or adenocarcinoma in situ (arrow). b) Follow-up HRCT after 5 years demonstrates increase in overall nodule size with a new <5 mm solid component, which suggests progression to minimally invasive adenocarcinoma. c) Fiducial marker was placed prior to surgical resection. d, e) Progression of minimally invasive adenocarcinoma to invasive adenocarcinoma. Note, 27 mm part-solid nodule in the right upper lobe with central <5 mm solid component (arrowhead) suggestive of minimally invasive adenocarcinoma and adjacent subcentimetre ground-glass nodule (arrow) presumed atypical adenomatous hyperplasia or inflammatory (d). e) HRCT at 2 years follow-up shows increase in size and attenuation of the part-solid nodule and growth of its solid stromal component, consistent with progression to invasive adenocarcinoma. The previously seen small, pure ground-glass nodule, which was likely inflammatory, has resolved.
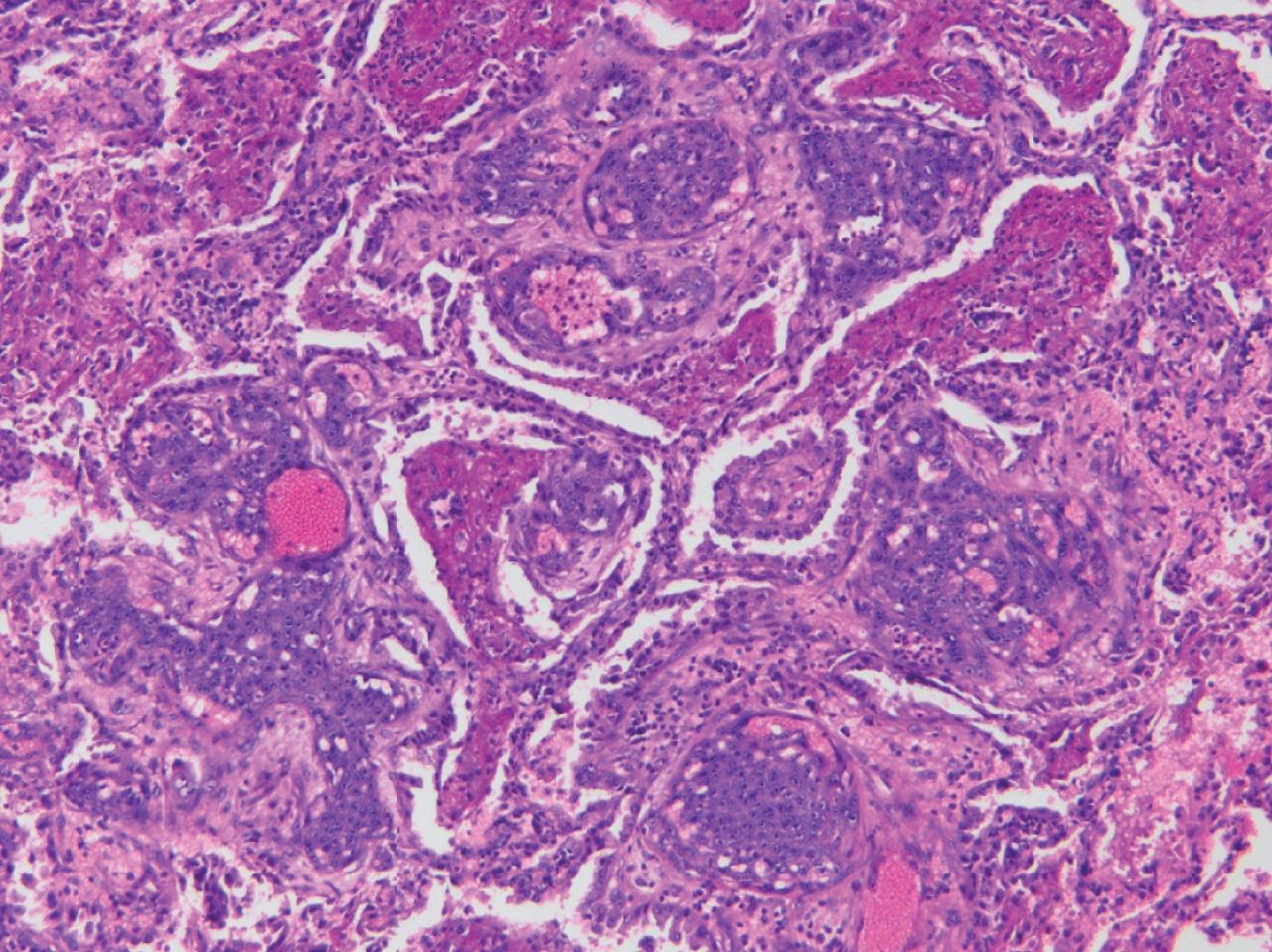
AIS is a malignant, pre-invasive malignancy that is further classified into three types: mucinous, non-mucinous and mixed. It tends to arise from the distal airways or alveoli, either from glandular cells or Type 2 pneumocytes. AIS grows along the alveolar septa (“lepidic growth pattern”) and does not invade into the surrounding airspaces, stroma, blood vessels or pleura (figure 20). Patients are usually asymptomatic, and these lesions are identified as incidental nodules on routine imaging.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
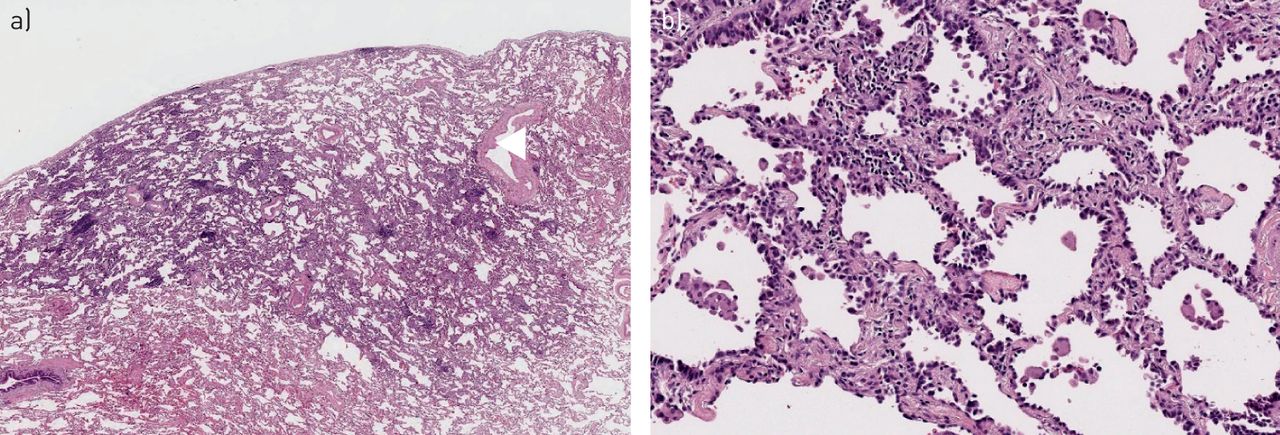
Adenocarcinoma in situ. a) Malignant glandular epithelium present on alveolar surface without evidence of invasion into the underlying lung (haematoxylin and eosin; 12.5×). b) Malignant glandular epithelium characterised by hyperchromatic nuclei with mild to moderate cytological atypia covers the alveolar surface (haematoxylin and eosin; 100×).
The incidence of true AIS is thought to be much less due to the more restrictive inclusion criteria proposed by the WHO. The incidence is higher in females, Asians and nonsmokers.
The pathological diagnosis of AIS is a nodule comprised of malignant glandular epithelium with a lepidic growth pattern, ≤3 mm, without evidence of invasion into the surrounding lung. This requires complete excision of the tumour and microscopic examination of the entire nodule to confirm there is no evidence of invasion into the surrounding lung. Hence, a diagnosis of AIS cannot be made with fine-needle aspiration or core biopsy [93].
AIS has an excellent prognosis if completely surgically resected with a cumulative incidence of recurrence of 0% at 5 years [94]. Patients can be followed using radiographic surveillance, though complete surgical excision should be done for definitive treatment.
Role of positron emission tomography imaging in the diagnosis of multiple lung nodules
The American College of Chest Physicians suggests a role for positron emission tomography (PET) in the evaluation of indeterminate lung nodules [95]. It is best suited to characterise indeterminate solid nodules >8 mm in patients with low-to-moderate probability of malignancy [96]. High-risk patients should undergo biopsy and very low-risk patients should undergo CT surveillance. The size of nodules is a major deciding factor for PET and most existing PET scanners have a spatial resolution of 8–10 mm (smaller nodules like minute pulmonary meningothelial-like nodules and DIPNECH may not be detected). Lung adenocarcinoma and carcinoid tend to have low to intermediate uptake, while lymphoproliferative and vascular tumours show high fluorodeoxyglucose (FDG) uptake (PET positive), though FDG uptakes can vary and should not be used as a surrogate for tissue sampling.
Conclusion
Several rare primary lung neoplasms may present as multiple synchronous indeterminate lung nodules on chest CT. These are often not included in the initial differential diagnoses and merit increased awareness among radiologists, pathologists and pulmonologists, as well as a multidisciplinary team approach.
Footnotes
Published online 2 Sept, 2020; republished 16 Sept, 2020 with amendments to author name.
Provenance: Submitted article, peer reviewed
Author contributions: S. Ghosh, A.C. Mehta, S. Abuqayyas, S. Raju and C. Faver contributed to the content of this manuscript and to the approval of the final version to be published.
Conflict of interest: S. Ghosh has nothing to disclose.
Conflict of interest: A.C. Mehta has nothing to disclose.
Conflict of interest: S. Abuqayyas has nothing to disclose.
Conflict of interest: S. Raju has nothing to disclose.
Conflict of interest: C. Farver has nothing to disclose.
- Received November 18, 2019.
- Accepted March 8, 2020.
- Copyright ©ERS 2020.
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










