





Abstract
The lung is a mechanically active organ, but uncontrolled or excessive mechanical forces disrupt normal lung function and can contribute to the development of disease. In asthma, bronchoconstriction leads to airway narrowing and airway wall buckling. A growing body of evidence suggests that pathological mechanical forces induced by airway buckling alone can perpetuate disease processes in asthma. Here, we review the data obtained from a variety of experimental models, including in vitro, ex vivo and in vivo approaches, which have been used to study the impact of mechanical forces in asthma pathogenesis. We review the evidence showing that mechanical compression alters the biological and biophysical properties of the airway epithelium, including activation of the epidermal growth factor receptor pathway, overproduction of asthma-associated mediators, goblet cell hyperplasia, and a phase transition of epithelium from a static jammed phase to a mobile unjammed phase. We also define questions regarding the impact of mechanical forces on the pathology of asthma, with a focus on known triggers of asthma exacerbations such as viral infection.
Abstract
Bronchoconstriction in asthma is not only a symptom but is also a disease modifier. Targeting downstream mediators of mechanical force induction may provide opportunities for novel therapies for asthma and its exacerbations. http://bit.ly/2UktQDj
Introduction
The lung is a mechanically active organ; in utero, lung development is dependent on the presence of controlled mechanical forces [1–5] and, following birth, lung movement continues unabated until death. This ever moving and changing nature of the lungs has been apparent since before Heraclitus, although it has only recently become apparent that alterations in the mechanical forces present in the lungs can generate and propagate lung disease [6–10].
During lung growth, mechanical forces are critical to alveologenesis and branching morphogenesis [1–5]. Even in utero, the fetal lung is on the move; breathing movements generated by diaphragmatic contractions leading to cyclic mechanical stretch are fundamental to maintaining airway luminal liquid volume, facilitating cellular proliferation and driving fetal lung expansion [11, 12]. At birth, fluid within the alveoli clears as the lung is aerated and expanded, developing alveolar surface tension [4], and from that moment lung elastic recoil is balanced by negative intrapleural pressure; a combination of mechanical forces at this point leads to postpartum lung maturation [13, 14]. Absence of stretch and mechanical force generation within the lung leads to abnormal alveolar growth and differentiation, especially of type II alveolar epithelial cells, reducing surfactant release and impairing gas exchange [15, 16].
For respiration to take place, the mature lung has to dynamically inflate and deflate; these repeated cycles of inflation and deflation generate mechanical forces, which are required for normal lung function. While maintenance of normal physiological force is essential for respiration, abnormal or uncontrolled mechanical forces generated in the airways can compromise normal lung function and result in disease pathogenesis [10, 17]. Although the impact of mechanical forces is becoming more understood in a variety of lung diseases, this review focuses on their impact in asthma.
Uncontrolled mechanical forces in lung pathophysiology
Airway narrowing (bronchoconstriction) occurs in asthma due to the contraction of airway smooth muscle (ASM) [18], leading to many asthma symptoms, including shortness of breath, chest tightness, cough and wheeze [19, 20]. As a result of bronchoconstriction, the airway wall in asthmatics had been thought to fold, buckle and form rosette patterns, and although this had been modelled extensively in silico and in vitro [6, 21, 22] it has only recently been observed in vivo [23]. Using optical coherence tomography (OCT) via a fibreoptic bronchoscope in human asthmatic and non-asthmatic volunteers, Adams et al. [23] documented airway folding following bronchoconstriction induced by experimental segmental allergen instillation, not only immediately following allergen instillation, but also at later time-points. The airway folding demonstrated by OCT showed remarkably similar airway changes to those previously predicted by in silico modelling and those found in pathological specimens of airways from fatal episodes of asthma [22, 24]. As the airway wall folds, the epithelium experiences multiple mechanical forces, including compression (as adjacent cells push against each other), stretch (of cells at the apex of epithelial folds) and shear stress (as air velocity increases in conjunction with a reduction in airway diameter) [5, 22]. Of these forces, compression has been most studied using a number of in vitro models which allow cultured epithelial cells to be subjected to compressive stress mimicking the mechanical effect of bronchoconstriction [6, 25–28]. These studies have demonstrated that mechanical forces can contribute to changes within the epithelial and other structural cells of the airway, changes that are consistent with features seen in asthma despite the absence of inflammatory stimuli. Application of mechanical compression in vitro recapitulates key features of asthmatic airway remodelling, including goblet cell hyperplasia, collagen deposition [6, 9, 29], and hyperplasia and hypercontraction of ASM cells [30], as well as inducing in normal cells a gene signature normally associated with asthmatic cells [31]. These studies offer a potential alternate or parallel pathway of asthma pathogenesis outside the accepted dogma of inflammation of a variety of subtypes (i.e. T-helper (Th)2 high, Th2 low etc.) driving disease progression, whereas bronchoconstriction results in a cascade of disease-initiating or disease-amplifying events, which may act in concert with, or independent of, inflammatory cells [32–35].
Modelling mechanical compression
Although the magnitude of the mechanical forces exerted on the epithelium during bronchoconstriction cannot be directly measured, it can be calculated using a finite element method which integrates both material properties and geometry of the airways [22]. This model uses two layers to describe the airway, representing an inner basement membrane combined with epithelium and an outer ASM layer (figure 1a). Constriction of the ASM layer leads to inner layer folding and, by increasing the thickness of the inner basement layer, mimicking the thickened sub-epithelial collagen in asthmatic airways, results in larger epithelial folds and airway occlusion [22]. As mentioned previously, this folding and occlusion appear similar to that seen both in airways of patients who suffer fatal asthma attacks (figure 1b) [24], and recently observed to a lesser degree via OCT (figure 1c) [23].

a) Schematic diagram showing buckling of airways with epithelial folding. Reproduced and modified from [22] with permission. b) Pathological specimen from fatal asthma with epithelial folding and luminal closure. Reproduced and modified from [24] with permission. c) Bronchoconstriction-induced airway wall folding following allergen challenge observed using optical coherence tomography. Reproduced and modified from [23] with permission.
The estimated apical pressure that is exerted on airway epithelial cells during bronchoconstriction is approximately 30 cmH2O pressure, which is an order of magnitude higher than transpulmonary pressure during tidal breathing (∼5 cmH2O) [22, 36]. Studies using canine airways also reported a similar transmural bronchial pressure in both large and small airways following acetylcholine-induced bronchoconstriction [37] and, as epithelial responses to apical compression plateau at 30 cmH2O pressure [6, 29], this force has been widely used to recapitulate the effects of asthmatic bronchoconstriction in vitro [7–9, 28, 38–41].
Experimental models for mechanical compression
In vitro model: air–liquid interface cultures
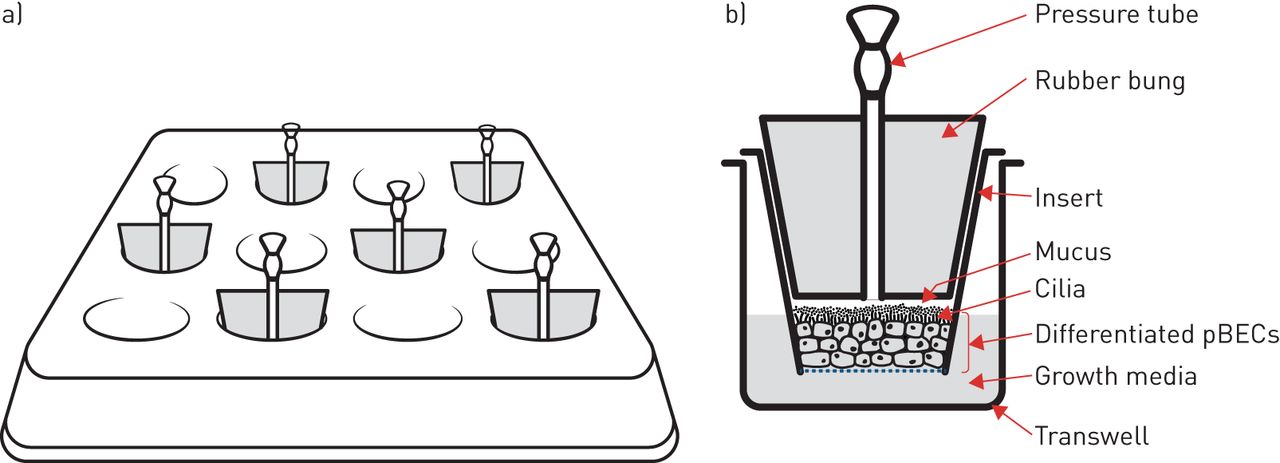
In vitro investigation of airway mechanical forces has primarily utilised human primary bronchial epithelial cells (pBECs) cultured at the air–liquid interface. Here, pBECs differentiate from a monolayer of basal cells to a pseudostratified structure containing goblet- and ciliated-cells to closely replicate the mature bronchial epithelium [42–44]. This well-differentiated airway epithelium is then exposed to a transcellular pressure gradient up to 30 cmH2O pressure (figure 2) to recapitulate the forces experienced during bronchoconstriction [22]. In addition to this apical stress model, another, more technically complex apparatus utilising lateral compressive stress has reported similar responses to apical compression [26, 27].
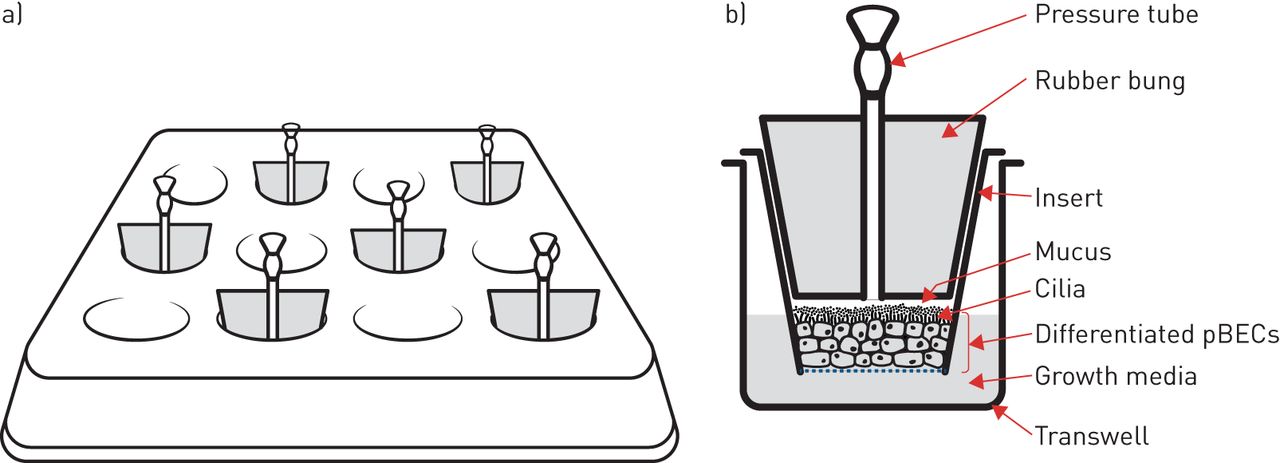
Apical compression system in vitro. a) Schematic of apical compression set-up on a 12-well transwell plate. b) Schematic of a frontal plane section of a single transwell insert enclosed by a bung with a pressure tube. pBEC: primary bronchial epithelial cell.
Mechanical compression of well-differentiated pBECs induces the release of growth factors into the lateral intercellular space between cells [38]. At the same time, mechanical compression collapses the lateral intercellular space without altering cell volume, this reduction in the volume of the lateral intercellular space results in an increase in the local concentration of epidermal growth factor (EGF) with consequent activation of the EGF receptor (EGFR) [38]. This pathway appears to mediate some of the changes, such as mucus secretion, that occur following modelled bronchoconstriction.
Ex vivo: precision-cut lung slices and whole lung perfusion models
In addition to pBEC monolayer in vitro models, epithelial compression during bronchoconstriction has been studied ex vivo using precision-cut lung slices (PCLS) and whole lung perfusion models, providing additional evidence in support of in vitro data. In PCLS, normal tissue architecture is maintained along with cellular communication between different cell types, albeit in thin lung slices; these slices can be generated from many animals, including humans [45–52]. Airways in PCLS exhibit bronchoconstriction and formation of a buckled epithelium following stimulation with agents such as methacholine or histamine [53] in a similar manner to isolated whole lung perfusion, where isolated lungs are generally perfused with a methacholine solution by puncturing the lung apex with multiple holes [38]. Though only a handful of studies have used PCLS or whole lung perfusion to examine the effects of bronchoconstriction, these techniques provide similar results to those from pBEC in vitro work (as discussed in the section Mechanical compression activates disease-associated pathways in the airway epithelium), supporting their conclusions.
In vivo: bronchoprovocation/allergen challenge models
Despite data from well-established in vitro and ex vivo models of airway compressive stress, until recently the question remained whether bronchoconstriction-induced epithelial buckling and mechanical stress actually occur in the human airway during an asthma exacerbation. It appears that these changes are likely to occur in real-world asthma; in addition to real-time OCT data [23], biopsies from patients with mild asthma undergoing repeated bronchoconstriction show pathological airway remodelling (structural changes) such as goblet cell hyperplasia and increased collagen deposition, in the absence of additional airway inflammation [10]. These remodelling changes were prevented by the administration of bronchodilators and inhaled corticosteroids during airway challenges [54, 55]. The threshold or number of bronchoconstriciton events required to induce any long-term changes in the airway is unknown. Participants with mild asthma in clinical studies who underwent a variety of bronchoprovocation tests, over a period of in some cases several years, did not show any unexpected fall in their forced expiratory volume in 1 s [56]. This could be that the frequency of challenge was too low, and the background asthma too mild to induce long-term changes, or that the changes observed after repeated allergen or methacholine challenge are transient, especially in otherwise well-controlled patients.
Data from animal models have also contradicted some of the human data. For example, mice exposed to a 30 min methacholine challenge at 48 h intervals over 6 weeks demonstrated airway remodelling only in terms of goblet cell hyperplasia and some mild macrophage-induced airway inflammatory response [57]. There was no effect on respiratory mechanics or the contractile capacity of ASM. These species differences mean that animal data must be interpreted with care when investigating the role of mechanical forces in human airway diseases.
Mechanical compression activates disease-associated pathways in the airway epithelium
Mechanical compression mimicking the mechanical environment of constricted airways recapitulates features of asthmatic airways, in addition to the activation of multiple signalling pathways leading to the production of asthma-associated mediators (figure 3). These pathways and mediators include the activation of EGFR, transforming growth factor (TGF)-β protein release, as well as procoagulant factors, and less well-understood compounds including YKL-40 and maspin.

{kind=link}
{kind=link}
{kind=link}
Schematic image of the effects of airway mechanical compression on airway epithelial cells and remodelling. Dotted lines indicate the mediator's effect on airway remodelling components. ASM: airway smooth muscle; EGFR: epidermal growth factor receptor; EGR-1: early growth response-1; HB-EGF: heparin-binding epidermal growth factor-like growth factor; PAI-1: plasminogen activator inhibitor-1; PAR: plasminogen activator receptor; TGF-α: transforming growth factor-α; TGF-β2: transforming growth factor-β2; uPA: urokinase plasminogen activator.
EGFR activation
Mechanical compression of well-differentiated epithelial cells results in the activation of EGFR and downstream signalling pathways [38, 58, 59]. For example, compressive stress induces phosphorylation of extracellular-regulated kinase (ERK) which upregulates heparin-binding EGF-like growth factor (HB-EGF) gene expression [60]; HB-EGF induction is EGFR-dependent, being activated via an autocrine positive feedback mechanism [41]. In mouse tracheal epithelial cells, activation of EGFR is mediated by EGF and induces ERK and protein kinase B (Akt) phosphorylation while this activation further induces the expression of the EGFR family of ligands, such as HB-EGF, TGF-α, amphiregulin, epiregulin and betacellulin. These findings are supported by in vivo studies where methacholine-induced airway constriction of mouse lungs also leads to the release of EGFR family ligands, with later data demonstrating that mechanical activation of EGFR requires active TNF-α converting enzyme (TACE) [38, 59].
TGF-β
TGF-β is a pleotropic cytokine with three different mammalian isoforms (TGF-β1–3) which plays an important role in regulating inflammation, cell growth and differentiation [61]. All three isoforms are expressed in the airways, but localise to specific cell types: TGF-β1: ASM, fibroblasts and endothelial cells; TGF-β2: airway epithelial cells; and TGF-β3: macrophages [62–68]. In many studies of asthma, TGF-β expression is elevated in the airways in comparison to healthy controls and induces airway hyperresponsiveness, as well as airway remodelling by increasing the number of goblet cells, mucus secretion and thickened sub-epithelial basement membrane by collagen or extracellular matrix (ECM) deposition [67, 69]. In contrast, some studies highlight no difference in isoform-specific TGF-β expression in asthma and healthy controls, especially with respect to TGF-β1 expression [66, 68, 70, 71]. These observed differences may be due to dichotomous expression of TGF-β1 to TGF-β2 and TGF-β3 or potentially the specific asthma phenotype or techniques used to detect TGF-β1 expression.
Despite these caveats, it has been shown that following mechanical compression TGF-β is released from in vitro pBEC cultures and plays a pivotal role in goblet cell hyperplasia, sub-epithelial collagen deposition and fibrosis [7, 9, 10]. Initial work in differentiated rat tracheal epithelial cells demonstrated increased TGF-β1 gene expression in response to mechanical compression in a time- and pressure-dependent manner. In well-differentiated human pBECs, TGF-β2, but not TGF-β1, protein expression was increased in response to compression [7]. pBECs derived from patients with asthma released more TGF-β compared with healthy controls following apical mechanical compression [28].
In guinea pig PCLS, methacholine-induced bronchoconstriction-induced biologically active TGF-β release from airway epithelial cells, promoting expression of ASM contractile proteins (smooth muscle-α-actin, smooth muscle-myosin, calponin) [50]. The TGF-β response was inhibited by preventing bronchoconstriction using latrunculin A to prevent actin polymerisation, and was also reduced by inhibiting TGF-β receptor kinase or phosphodiesterase-4 and also by the administration of long-acting muscarinic receptor antagonists [50, 72]. Finally, in human studies, repeated bronchoconstriction of mild asthmatic patients showed elevated epithelial TGF-β expression compared with saline-treated individuals, along with changes in other mediators in bronchial biopsies [10]. Together, these studies demonstrate that TGF-β in airway epithelial cells is responsive to mechanical compression associated with bronchoconstriction as well as being linked to airway remodelling and airway hyperresponsiveness [73], and may provide a mechanistic connection between epithelial compression and asthma pathogenesis.
Procoagulant factors
Fibrin degradation products and the coagulation cascade are activated in the days preceding admission to hospital for an asthma exacerbation, and the degree of activation is associated with length of stay for those exacerbations [74, 75]. Coagulation cascades involving the activation of plasminogen systems, including urokinase plasminogen activator receptor, urokinase plasminogen activator, plasminogen activator inhibitor-1 and amphiregulin are upregulated after epithelial cell compression [8]. Tissue factor, a well-recognised coagulation initiator, is expressed in human airway epithelial cells; both gene expression and protein release are increased following compression [39]. Tissue factor protein is released via extracellular vesicles, and this release is augmented by interleukin (IL)-13, a type-2 cytokine, elevated in allergic asthma [76]. Tissue factor is expressed in pre-clinical models of allergic airways disease and is detected in bronchoalveolar lavage fluid from ovalbumin-challenged allergic mice. At baseline, tissue factor levels are higher in asthmatics compared with healthy controls and increase further in asthma in response to allergen challenge [39, 76]. It may be that bronchoconstriction, in combination with the underlying state of airway inflammation, directly leads to clotting cascade activation. It also appears that bronchoconstriction interacts with other disease processes in asthma and this interaction could provide a target for novel interventions.
YKL-40 and maspin
Genome-wide association studies have associated CHI3L1, the gene which encodes the YKL-40 protein, with increased asthma prevalence or susceptibility in multiple populations [77–80]. Higher expression of CHI3L1 in the lung correlates with higher concentrations of YKL-40 in serum and bronchoalveolar lavage fluid [81], which are themselves associated with reduced lung function [77]. More recent evidence suggested that YKL-40 plays a role in airway remodelling, including airway angiogenesis [82, 83] and ASM proliferation [84]. Compression of pBECs induces YKL-40 protein secretion, mediated through the activation of the EGFR-ERK pathway [40], although the relationship between CHI3L1, as an asthma susceptibility gene, and the secretion of YKL-40 protein from compressed airway epithelial cells is not yet clear. In addition to YKL-40, another protein which contributes to tissue remodelling, maspin, is also released from epithelial cells following apical compression or allergen exposure [85]. Maspin, a serine protease inhibitor, originally identified as a tumour suppressor protein in human mammary epithelial cells, contributes to tissue remodelling by inhibition of collagen degradation and alteration in cell–cell interactions [86–88]. The interaction of bronchoconstriction with genetic susceptibility to disease and proteins responsible for tissue remodelling will be a novel area for investigation.
Mechanical compression induces airway remodelling
Remodelling of the airways in asthma is characterised by increased goblet cell number, mucus hypersecretion, sub-epithelial collagen deposition and an increased ASM mass [18, 89, 90]. The driving factors behind, and clinical effect of, these changes are not clear, but are thought to be deleterious; mucus secretion leads to airway plugging and closure, collagen deposition contributes to fixed airway obstruction, and ASM hypertrophy contributes to dynamic airway narrowing and closure.
Goblet cell hyperplasia and MUC5AC protein expression
Goblet cells are non-ciliated secretory airway epithelial cells which secrete glycoprotein rich mucus into the airway lumen. This mucus forms a barrier to environmental insults and increases the airway's ability to clear pathogens or pollutants by mucociliary action [91–93]. In asthma, goblet cells increase in number and secrete more mucus; these increases are associated with an increase in asthma morbidity and mortality [89, 94–96]. This increased goblet cell number and mucus production is associated with increased Th2 cytokines (IL-4, IL-5 and IL-13) and human neutrophil elastase in the airways [97–99]. Goblet cell numbers are further increased in vivo following repeated experimentally induced bronchoconstriction in mild asthmatics and in vitro by apical compression of pBECs for only 1 h a day for 14 days [9, 10]. Compression applied in vitro leads to an increase in MUC5AC protein expression in addition to goblet cell hyperplasia which is decreased following pre-treatment of pBECs with an EGFR kinase inhibitor. EGFR inhibition in combination with a TGF-β2 neutralising antibody completely prevents this effect [9]. Although the mechanisms of goblet cell hyperplasia are not well understood, and may include the Notch3 pathway [100], this phenomenon has been shown to occur in the healthy mouse lung following repeated methacholine challenge [57].
Sub-epithelial collagen deposition
In addition to mucus production, thickening of ECM is another feature of asthmatic airway remodelling [69, 101]. ECM deposition occurs at the basolateral surface of the bronchial epithelium and consists, at least in part, of collagens (type I, III and IV) as well as fibronectin [69, 101–103]; asthmatic airways have measured ECM thicknesses of ∼9 µm in comparison to 5–6 µm for non-asthmatics [104]. Compressive stress promotes airway remodelling by inducing the deposition of sub-epithelial collagen and fibronectin [26, 29], this appears to be a result of epithelial cells responding to mechanical stress and releasing cellular factors which induce proliferation of fibroblasts and increased production of collagens (types III and V) [29]. In addition to the ECM deposition observed in this model, there was also an increase in matrix metalloproteinase-2 and -9 production [27, 29].
ASM proliferation and contraction
The ASM in asthmatic airways is characterised by increased mass and increased responsiveness to stimuli, which together result in excessive contraction [105–107]. Although the mechanism via which ASM becomes more responsive to contractile stimuli is unclear; it has been demonstrated that airway epithelial cells can augment ASM contraction and proliferation [108–110] via mediators such as endothelin (ET)-1 and YKL-40 [84, 111, 112]. Both these mediators (ET-1 and YKL-40) have been shown to be increased following the application of apical mechanical stress on epithelial cells in culture [7, 40] with condition media from those epithelial cells inducing ASM hypertrophy which was blocked by ET-1 antagonists, while ASM proliferation was also increased, but through an ET-1 independent mechanism [30]. These data indicate that mechanical compression of airway epithelial cells can elicit a response in the ASM and potentially generate a positive feedback loop that may be a critical therapeutic target for arresting asthma progression.
Mechanical compression induces epithelial cell unjamming
Collective cellular migration
It is widely recognised that airway epithelial cells migrate during wound repair; however, recently it has also been demonstrated that unwounded well-differentiated airway epithelial cells also can migrate in vitro [113]. Normally, in vitro, airway epithelial cells migrate relative to each other within the culture environment during their growth and differentiation phase, “locking” into a fixed arrangement as they reach maturity at the air–liquid interface [114]. Application of mechanical compression to well-differentiated “fixed” cells leads the epithelium to undergo a striking phase change, where the cells become motile and begin to flow collectively. This change from a static and solid-like jammed state to a fluid-like unjammed state has been defined as an unjamming transition [113, 114].
Cell jamming and cell shape
Jamming traditionally refers to a physical phenomenon whereby disordered materials, such as particulates, foams or colloids can acquire structural rigidity without the formation of crystal structure [115]. Jamming has been thoroughly investigated in inert materials as a function of density [116] but, recently, Bi et al. [117] sought to expand this paradigm to epithelial cellular systems where density may not change, but the cells within the system could exist in a solid-like or fluid-like state [117]. Their theory predicted that tissue behaviour would be determined by epithelial cell shape where a critical threshold exists in which cells “below” this cell shape threshold (more rounded or cobblestone-like) would display solid-like behaviour, whereas cells “above” this cell shape threshold (more elongated or anisotropic) would display fluid-like behaviour. This prediction was proved by Park and co-workers [113, 114], who showed that cells in the asthmatic epithelium jammed and unjammed dependent on cell shape. It has now been further shown that cell shapes and cell shape variability in a large range of epithelial systems are mutually constrained through a geometric relationship that is connected to jamming [118].
Taken together, this work has demonstrated that in the solid-like jammed phase, local rearrangements of cells do not occur due to the large energy barrier, whereas in the fluid-like unjammed phase, cellular rearrangements are possible or even favoured due to the vanished energy barrier. Epithelial cells exist in homeostatic conditions close to a jamming transition but can unjam, and therefore remodel, in response to many internal or external cues [114, 119].
Although the impact of collective cellular migration has not been established in vivo, previously published papers suggest a potential link between unjamming and asthma pathogenesis. For example, pBECs cultured from asthmatic donors show a delayed transition from the unjammed to jammed phase during maturation in air–liquid interface culture; a transition from a jammed to an unjammed phase is provoked by mechanical compression mimicking bronchoconstriction [113]. As epithelial cell unjamming is accompanied by prominent cell shape changes; these changes of cell shape infer changes in mechanics within the cells or in their local environment. Altered cellular mechanics through cell shape changes impact many cellular functions, including secretion of proteins [120, 121], relocation or activation of transcription factors, changes in downstream gene expression [122–125], and cell fate decisions [126]. As changes in cell shape and cell mechanics directly link to cell and tissue function [127], it is possible that compression and disease are linked through unjamming-related changes in cell shape.
As the study of cellular unjamming transitions is a relatively new field, the question of whether it is an epiphenomena or a factor in health or disease remains despite its demonstration in a variety of systems. Recent studies reveal the occurrence of this unjamming phenomenon during the development in a variety of systems. These include during ventral furrow formation during gastrulation in Drosophila [118], during elongation of the vertebrate body axis in the embryonic zebrafish [128], and during airway epithelial branching in the embryonic avian lung [129].
Although difficult to assess, the presence or absence of cellular jamming not only in asthma but also in other respiratory diseases in vivo could be assessed by an assessment of cellular shape. These approaches should be possible at bronchoscopy, either via biopsy, or perhaps by OCT, though were not investigated by Adams et al. [23] in their recent publication; this may establish a causal link between the unjammed phase of epithelium and disease. Once established, the role of asthma mediators and medications could be assessed for their influence on the unjamming transition.
Mechanical compression may impair epithelial anti-viral immunity
One of the fundamental roles of the airway epithelium is to protect against viral infection, including an innate immune response to viruses [130]. Patients with severe asthma are commonly found to exhibit reduced barrier function and as such have a greater susceptibility to lower respiratory tract illness particularly triggered by rhinovirus (RV) infections and have reduced epithelial defence responses [131–133]. Asthmatic airway epithelial cells also appear to have impaired innate immune responses following RV infection, including reduced production of interferons (IFNs) which often results in increased and prolonged RV replication [134–136]. This increased RV replication may translate to more severe viral infections in asthmatics [137].
Although there appears to be an intrinsic deficiency of IFN production from epithelial cells of asthmatics, this deficiency can be augmented by local mediators produced from epithelial cells in response to viral infection [138, 139]. In monolayer culture of epithelial cells from non-asthmatics infected with RV, the addition of TGF-β2 decreased release of IFN protein and increased viral replication; these effects were reversed by TGF-β neutralising antibodies. In the same study, cells cultured from asthmatics, where endogenous TGF-β2 concentrations are higher, also showed decreased IFN protein release, which was increased by the addition of anti-TGF-β antibody [138]. Also, viral infection may directly activate EGFR, leading to a reduction in IFN production from airway epithelial cells [139]. These data suggest that TGF-β and EGFR activation may further worsen anti-viral innate immune deficiencies in asthma.
Airway mechanical forces may influence glucocorticosteroid sensitivity
Inhaled corticosteroids (ICS) are fundamental to the treatment of asthma [140]. ICS treatment supresses type-2 inflammation and contributes to asthma control and a reduction in airway hyperresponsiveness [141–143]. However, there are groups of asthmatics in whom ICS treatment is less effective, including patients with non-eosinophilic asthma and more severe disease [35, 144, 145]. Even in the corticosteroid sensitive eosinophilic asthma phenotype, approximately 10–20% of patients respond sub-optimally to these drugs [146]. The airway epithelium is central to ICS resistance, and increasing resistance to ICS has been demonstrated during bronchoconstriction and RV infection through several pathways [147–149]. RV infection also modifies ICS activity and impairs the ability of ICS to inhibit release of the pro-inflammatory cytokines IL-1β and CXCL8, by reducing translocation of the glucocorticoid recpetors to the nucleus [150]. As both RV infection and bronchoconstriction interfere with ICS function, it may be that relative inhaled glucocorticoid resistance during viral exacerbations of asthma could be secondary to an interaction between viral infection and mechanical forces within the airway.
Conclusion
Bronchoconstriction induces airway structural changes in asthma even in the absence of additional airway inflammation. Airway distortion has now been shown to persist for an extended period after the induction of airway narrowing and may lead to the progression of asthma via airway structural changes, diminished responses to viral infections or decreased efficacy of commonly used medications. Targeting bronchoconstriction as a disease modifier as well as a symptom of asthma could lead to better outcomes for our patients, and it is possible that downstream mediators upregulated by mechanical force induction may provide opportunities for novel therapies for asthma.
Footnotes
Published online 4 Aug, 2020; republished 19 Sept, 2020 with amendments to figure 2.
Provenance: Submitted article, peer reviewed
Conflict of interest: P.C Veerati has nothing to disclose.
Conflict of interest: J.A. Mitchel has nothing to disclose.
Conflict of interest: A.T. Reid has nothing to disclose.
Conflict of interest: D.A. Knight has nothing to disclose.
Conflict of interest: N.W. Bartlett has nothing to disclose.
Conflict of interest: J-A. Park has nothing to disclose.
Conflict of interest: C. Grainge has nothing to disclose.
- Received September 25, 2019.
- Accepted January 30, 2020.
- Copyright ©ERS 2020.
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References











Jump To
- Article
- Abstract
- Abstract
- Introduction
- Uncontrolled mechanical forces in lung pathophysiology
- Modelling mechanical compression
- Experimental models for mechanical compression
- Mechanical compression activates disease-associated pathways in the airway epithelium
- Mechanical compression induces airway remodelling
- Mechanical compression induces epithelial cell unjamming
- Mechanical compression may impair epithelial anti-viral immunity
- Conclusion
- Footnotes
- References
- Figures & Data
- Info & Metrics