





Abstract
Available bronchodilators can satisfy many of the needs of patients suffering from airway disorders, but they often do not relieve symptoms and their long-term use raises safety concerns. Therefore, there is interest in developing new classes that could help to overcome the limits that characterise the existing classes.
At least nine potential new classes of bronchodilators have been identified: 1) selective phosphodiesterase inhibitors; 2) bitter-taste receptor agonists; 3) E-prostanoid receptor 4 agonists; 4) Rho kinase inhibitors; 5) calcilytics; 6) agonists of peroxisome proliferator-activated receptor-γ; 7) agonists of relaxin receptor 1; 8) soluble guanylyl cyclase activators; and 9) pepducins. They are under consideration, but they are mostly in a preclinical phase and, consequently, we still do not know which classes will actually be developed for clinical use and whether it will be proven that a possible clinical benefit outweighs the impact of any adverse effect.
It is likely that if developed, these new classes may be a useful addition to, rather than a substitution of, the bronchodilator therapy currently used, in order to achieve further optimisation of bronchodilation.
Abstract
There is a real interest among researchers and the pharmaceutical industry in developing novel bronchodilators. There are several new opportunities; however, they are mostly in a preclinical phase. They could better optimise bronchodilation. http://bit.ly/2lW1q39
Introduction
While there has been significant progress over the years in the management of airway disorders, bronchodilator treatment, which remains a key element in the therapy of these disorders, is still stuck in the use of β2-adrenoceptor agonists, muscarinic acetylcholine receptor (mAChR) antagonists and xanthines, although there has been a gradual improvement in these classes of bronchodilators [1–3].
Certainly, available bronchodilators satisfy many of the needs of patients suffering from airway disorders, but they often do not relieve symptoms and their long-term use raises safety concerns. Therefore, there is interest in developing new classes that could help to overcome the limits that characterise the existing classes.
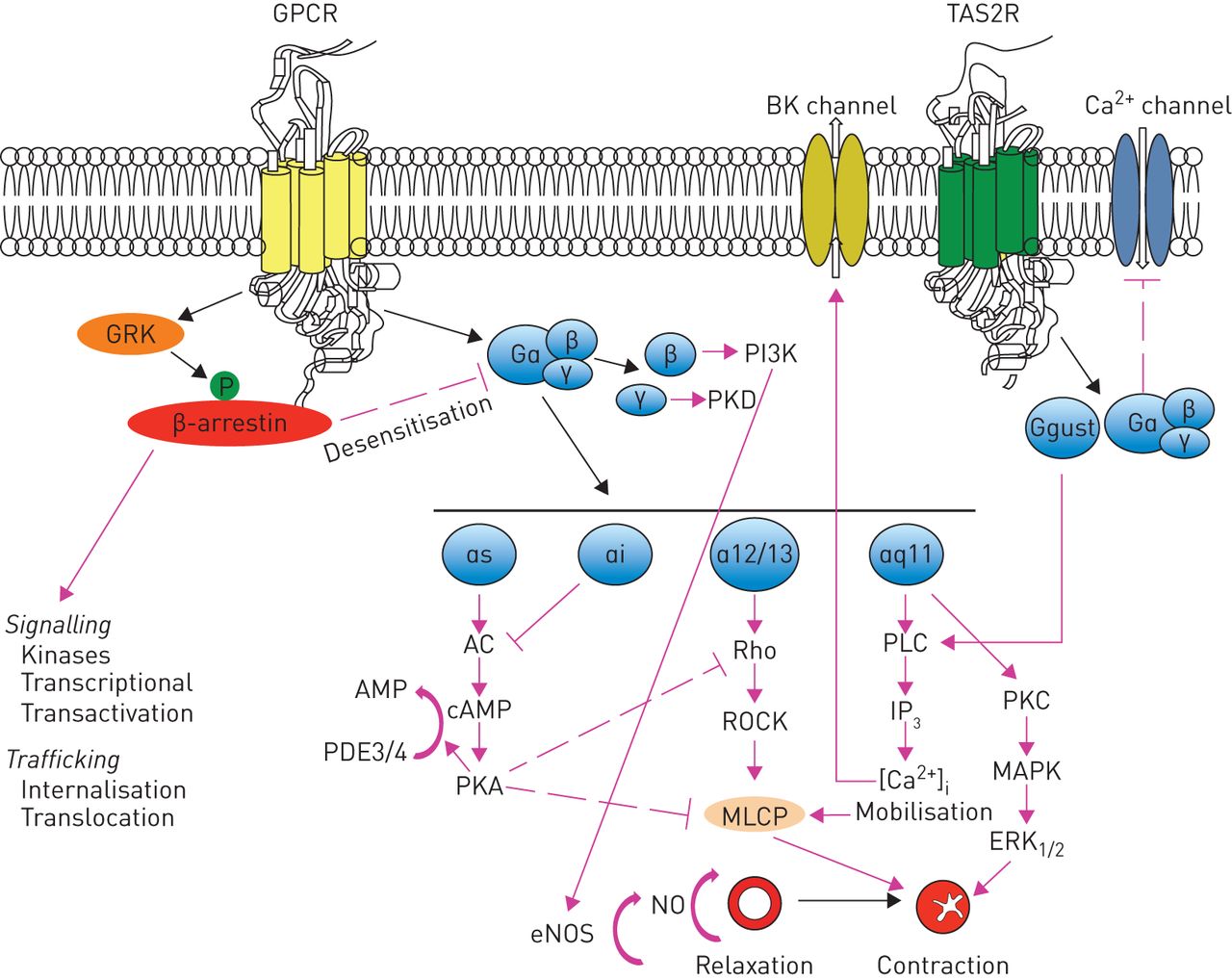
It has been repeatedly highlighted that novel classes of bronchodilators have proved difficult to develop, but in recent years, new targets for drugs to induce bronchodilation have been identified [4, 5]. In effect, the awareness that there are a large number of receptors in the G-protein-coupled receptor (GPCR) superfamily expressed in human airway smooth muscle (ASM) and extensive variability in structure because of alternative splicing leading to many receptor isoforms [6] (figure 1) led to identification of at least nine potential new classes of bronchodilators that are under consideration: 1) selective phosphodiesterase (PDE) inhibitors; 2) bitter-taste receptor agonists; 3) E-prostanoid (EP) receptor 4 agonists; 4) Rho kinase inhibitors; 5) calcilytics; 6) agonists of peroxisome proliferator-activated receptor (PPAR)-γ; 7) agonists of relaxin receptor 1; 8) soluble guanylyl cyclase activators; and 9) pepducins. There are other mechanisms for inducing bronchodilation, including the opening of potassium channels [2, 3, 5], but their importance has been questioned in human airways [7].
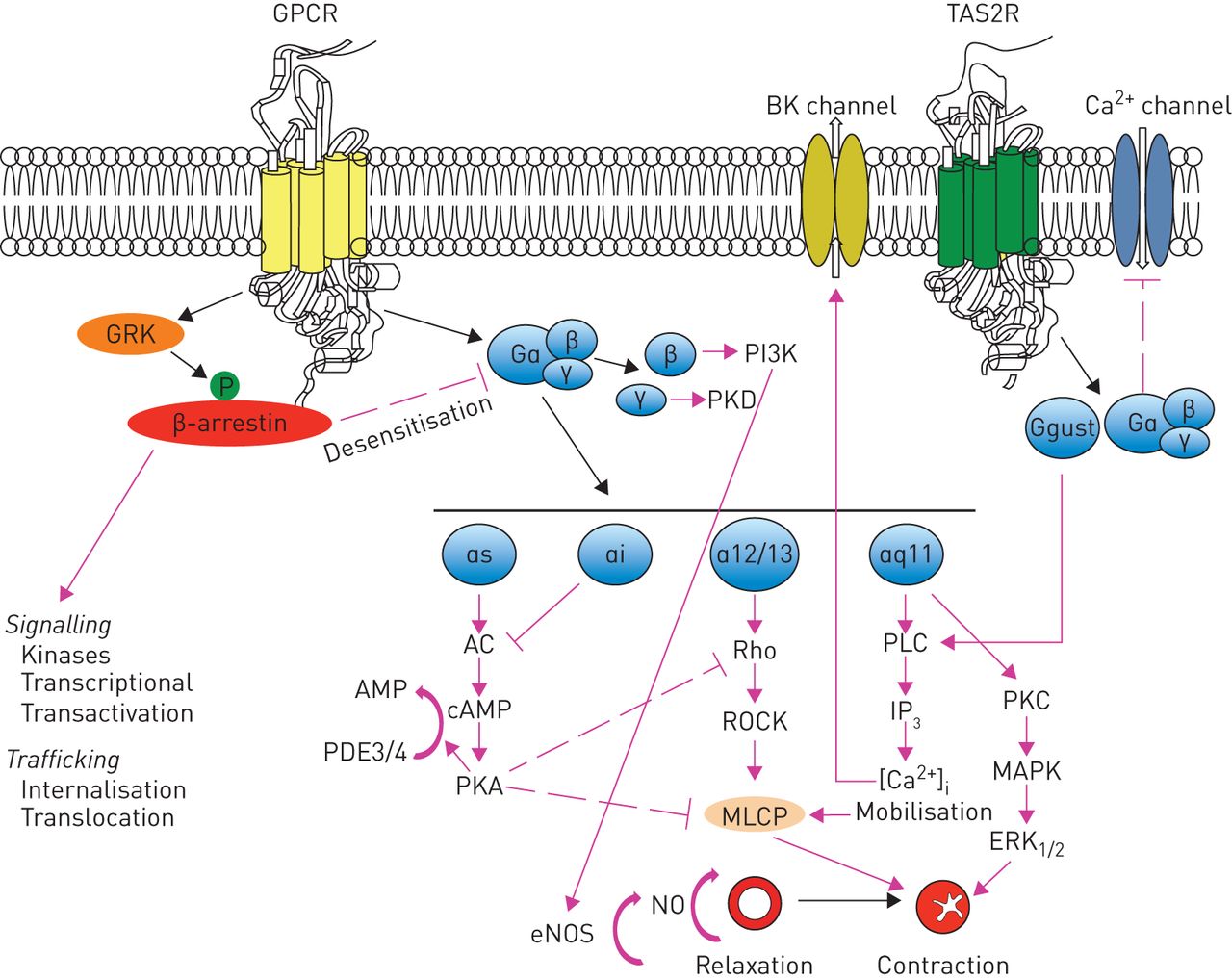
Synthesis of signalling and functional effects of various G-protein coupled receptors (GPCRs) that have been identified in airway smooth muscle cells. AC: adenylyl cyclase; cAMP: cyclic adenosine monophosphate; eNOS: endothelial nitric oxide synthase; ERK: extracellular signal-regulated kinase; GRK: GPCR kinase; IP3: inositol-3-phosphate; MAPK: p38 mitogen-activated protein kinase; MLCP: myosin light chain phosphatase; NO: nitric oxide; PDE: phosphodiesterase; PI3K: phosphatidylinositol 3-kinase; PKA: protein kinase A; PKC: protein kinase C; PKD: protein kinase D; PLC: phospholipase C; ROCK: Rho-associated coiled-coil-containing protein kinases; TAS2R: taste type 2 receptor.
The purpose of this narrative review is to summarise the existing evidence on emerging classes of bronchodilators, although many of the novel agents with bronchodilator properties included in our revision, mainly “bifunctional” molecules, also induce other significant effects that may be of interest in respiratory disease (e.g. anti-inflammatory, antifibrotic, etc.).
Selective PDE inhibitors
PDEs are not a real new target. In fact, roflumilast, which is a PDE4 inhibitor, has been available on the market for a long time for the treatment of COPD. However, we understand that different PDE isozymes selectively regulate cyclic adenosine monophosphate (cAMP) or cyclic guanosine monophosphate (cGMP) signalling in different subcellular microdomains, and individual PDEs are likely involved in specific locations at certain time-points based on different stimulations/activations (figure 2) [8, 9]. Furthermore, rather than involving a single PDE, we must consider the participation of multiple PDE variants in a complex signalling network involving central regulatory mechanisms [10, 11]. It is also likely that compounds that inhibit PDE4 enzymes together with a second PDE family could provide a therapeutic benefit at a concentration that does not cause emesis [12].

Potential sites of action of phosphodiesterase (PDE) inhibitors and soluble guanylyl cyclase (sGC) activators in airways smooth muscle and inflammatory cells. AC: adenylyl cyclase; (c)AMP: (cyclic) adenosine monophosphate; (c)GMP: (cyclic) guanosine monophosphate; GC: guanylyl cyclase; NO: nitric oxide; PKG: protein kinase G.
These concepts have led to the investigation of the PDE family as an important target in the treatment of airways disorders and to explore the possibility of developing drugs with the ability not only to inhibit PDEs not fully evaluated in the past, such as PDE8 [13] and PDE9 that seem to regulate bovine tracheal smooth muscle relaxation [14], but also to interact simultaneously with different PDEs [10, 11].
Dual PDE3/PDE4 inhibitors
The recognition that the PDE4 isoenzyme is the principal isoenzyme in the majority of inflammatory cells of importance in the pathogenesis of asthma and COPD, and that PDE3 is the predominant PDE isoenzyme in ASM and its inhibition produces ASM relaxation as well as enhancement of relaxation evoked by β2-adrenoceptor stimulation [15], has led to the development of drugs having dual inhibitory activity for both PDE3 and PDE4 in order to obtain both bronchodilator and anti-inflammatory activity in the same molecule [10, 11].
Several compounds that selectively inhibit PDE3 and PDE4, such as zardaverine, benzafentrine, tolafentrine and pumafentrine have been tested in human volunteers but none have progressed to the clinic presumably because of lack of efficacy or concerns about safety [9]. Ensifentrine (RPL554) is the only dual PDE3/PDE4 inhibitor under clinical development for the treatment of COPD, asthma and likely, cystic fibrosis [16]. However, it has been highlighted that its affinity for PDE3 is 3440-times higher than that for PDE4 (half maximal inhibitory concentration [IC50] for PDE3 0.43 nM and PDE4 1479 nM) [17], which suggests that it acts predominantly as a PDE3 inhibitor and therefore as a bronchodilator [9]. It is worth mentioning that there is preclinical and clinical documentation of a synergistic interaction between ensifentrine and β2-adrenoceptor agonists or mAChR antagonists [18–20], which suggests it could be used as an add-on agent. In any case, although there is concern that the blockage of PDE3 can cause adverse events, particularly in the cardiovascular system because this enzyme is also important in regulating the function of vascular smooth muscle and cardiac muscle, ensifentrine is a safe and well-tolerated drug [11].
Dual PDE4/PDE7 inhibitors
PDE4 and PDE7 are expressed in immune cells and specifically control intracellular levels of cAMP [21]. Simultaneous inhibition of PDE4 and PDE7 potentiates the effects observed by the mere inhibition of PDE4 or PDE7 [22] and suppresses airway hyperresponsiveness [23]. Nonetheless, greater anti-inflammatory activity of dual PDE4/PDE7 inhibitors over a PDE4 inhibitor alone has not been persuasively demonstrated [24] also because it has been shown that the inhibition of PDE7 is unable to influence proinflammatory cells per se but increases the inhibitory effect of other cAMP-elevating drugs [25]. Several PDE4/PDE7 inhibitors (e.g. YM-393059, T-2585, ASB16165 and BC54) have been examined. There is evidence that YM-393059, which inhibits PDE4 and PDE7 with IC50 values of 630 nM and 14 nM, exhibited some activity in preclinical models of inflammation [26], and T-2585, which inhibits PDE4 and PDE7 with IC50 values of 0.013 nM and 1.7 nM, blocked proliferation and cytokine release from T-cells when the highly selective PDE4 inhibitor, piclamilast had no effect [27]. ASB16165, which inhibits PDE4 and PDE7A with IC50 values of 2.1 nM and 15 nM, inhibited CD3/CD28-stimulated T-cell proliferation and cytokine release [28]. BC54 induced a larger anti-inflammatory effect on tumour necrosis factor-α production by macrophages and interleukin (IL)-2 production by T-lymphocytes as compared to rolipram alone or in combination with BRL50481 [29], and combined antisense inhibition of the expression of PDE4B, PDE4D and PDE7A produced a much greater anti-inflammatory effect than roflumilast alone in an in vivo mouse model of smoke-induced lung inflammation [22].
Because of these findings, there is still an interest in identifying new effective PDE4/PDE7 inhibitors mainly as anti-inflammatory agents, although the inhibition of PDE4 could influence ASM tone, and several compounds have been designed and synthesised, such as a series of butanehydrazide derivatives of purine-2,6-dione [30], a series of novel amide derivatives of 1,3-dimethyl-2,6-dioxopurin-7-yl-alkylcarboxylic acids that inhibit PDE4B/PDE7A activity and also block the human TRPA1 channel, although are more focused on the treatment of pain [31], and some newer substituted 1,3- thiazolidine-2,4-dione derivatives [32].
Dual PDE4/PDE5 inhibitors
Dual PDE4/PDE5 inhibitors could act at multiple levels in airways disorders, decreasing arterial pulmonary hypertension but also reducing lung inflammation and, probably, also remodelling and improving lung function [12]. There is preclinical evidence that combining selective inhibitors of PDE5 (tadalafil) and PDE4 (roflumilast) can suppress airway hyperresponsiveness and markers of inflammation, at least in the model of ovalbumin-induced eosinophilic inflammation, by increasing the intracellular levels of cAMP and cGMP via PDE4 and PDE5 inhibition [33].
LASSBio596, designed as a hybrid of thalidomide and aryl sulfonamide, was able to elicit an important anti-inflammatory function in a mouse model of Escherichia coli lipopolysaccharide-induced acute lung injury [34], lessening pulmonary inflammation and also to induce airspace enlargement and small-airway wall remodelling, thus expanding lung function in an animal model of elastase-induced emphysema [35]. However, it has not been clinically developed, probably because LASSBio-596 has a negative effect on cardiac development and function [36].
A small-molecule PDE4/PDE5 dual inhibitor named Compound A, which inhibits PDE4B, PDE4D and PDE5A with IC50 values of 6.7×10−10 M, <1×10−8 M and 6.6×10−10 M, respectively, has recently been tested in a rat model of monocrotaline-induced pulmonary hypertension [37]. It reduced profibrotic gene expression induced by transforming growth factor-β and suppressed right ventricular hypertrophy and C–C motif chemokine ligand 2 expression levels in the lung and ameliorated elevated plasma surfactant protein D.
Nonetheless, it has been suggested that there will be no clinical development of dual PDE4/PDE5 inhibitors because selective PDE5 inhibitors not only do not seem effective, but apparently they result in even worse gas exchange [38].
Bitter-taste receptor agonists
Bitter-taste or taste type 2 receptors (TAS2Rs) are a group of 25 proteins belonging to the GPCR superfamily that have been identified in the taste buds of the tongue and also in extraoral tissues, including respiratory epithelia and smooth muscle [39]. They are classified as class A GPCRs for their architecture and binding site location [40]. Three of these TAS2Rs, the subtypes 10, 14, and 31, are highly expressed in human ASM [41]. Their activation causes marked relaxation in intact airways of mice and humans, as well as isolated human ASM cells with an effect that is three-fold greater than β-agonists [42]. The mechanism by which this occurs remains uncertain. It has been suggested that it happens through a calcium-dependent, Gq-coupled mechanism [43] and subsequent activation of phospholipase-C (PLC)-β2 that mediates the synthesis of inositol-3-phosphate (IP3), which gates IP3 receptor type 3 (IP3R3) on the endoplasmic reticulum, causing Ca2+ release in the cytosol, leading to activation of large conductance KCa2+ channels (but also inhibition of Ca2+ sensitisation and actin polymerisation), membrane hyperpolarisation and bronchodilation (figure 1) [44]. Nevertheless, there is documentation in mice that ASM relaxation is caused by Gβγ subunits that inhibit L-type voltage-gated Ca2+ influx channels and thus reverse the increase in Ca2+ induced by bronchoconstrictors [45]. The activation of PDEs by the Gα subunit, which causes a decrease in the activity of protein kinase A (PKA) and consequently, a reduction in cAMP levels, is another possible mechanism. PKA phosphorylates IP3 receptor (IP3R), which becomes inactive by IP3 and, consequently, it inhibits IP3R-mediated Ca2+ release [46].
However, a dual mechanism of action has also been suggested for TAS2R agonists to relax ASM [47]. At high concentrations they would induce focal increases in the intracellular calcium ion concentration [Ca2+]i and membrane hyperpolarisation, leading to relaxation. When TAS2Rs are activated by lower concentrations of agonists under conditions of elevated [Ca2+] by various bronchoconstrictors, they would be able to inhibit the Ca2+ release induced by contractile agents in an as yet unknown manner; thus they would oppose membrane depolarisation and facilitate the relaxation of precontracted ASM.
The finding that TAS2R function in human ASM cells is independent of disease and inflammatory state is contrary to what is observed with β2-adrenoceptors, suggesting that TAS2R agonists may be useful bronchodilators as an adjunct to (or replacement for) β-adrenoceptor agonists [48].
Unfortunately, TAS2R ligands are chemically different, include ions, peptides, alkaloids, polyphenols, glucosinolates and others, and are agonists at most submicromolar activities, unlike typical class A GPCRs, which have many nanomolar ligands [48]. Furthermore, some TAS2Rs can detect several bitter compounds, whereas others detect one or only a few bitter compounds [49]. Even though many TAS2R ligands, including erythromycin, carisoprodol, flufenamic acid, dapsone, quinine, chloroquine, caffeine, azelastine and colchicine are already approved drugs, their potency against TAS2Rs is not enough for their repurposing to TAS2R-driven diseases [50], and many of them have pharmacological effects unrelated to the stimulation of TAS2Rs that cannot be ruled out as contributing to the beneficial effects seen on ASM [51].
There is currently very little clinical evidence supporting TAS2R agonists being a new class of bronchodilators for treating obstructive lung disease, and clearly there is a need to find further new bitter compounds to better understand the relationship between bitter receptors and bitter compounds. However, it has been highlighted that currently we still do not know anything about the properties of bitter compounds that could help in the search for TAS2R ligands [52].
Nevertheless, some research in the field is ongoing. Flufenamic acid, an anti-inflammatory and analgesic drug, is likely to be the most potent TAS2R agonist, although its activity towards its cognate bitter-taste receptor TAS2R14 requires concentrations of ≥100 nM [53]. However, slight modifications in its structure may radically reduce or even abolish TAS2R14 activity [54].
EP receptor 4 agonists
The beneficial effects of inhaled prostaglandin E2 (PGE2) on human airway calibre have been recognised for a long time [55, 56]. The biological actions of PGE2 are predominantly mediated by the activation of the four GPCRs, namely EP1, EP2, EP3 and EP4 [57]. Findings from studies that used human tissue clearly documented that PGE2-induced relaxation is mediated via the EP4 [58]. Activation of EP4 receptors stimulates adenylyl cyclase activity via the Gαs subunit resulting in an increase in cAMP (figure fig. 3) [57]. Therefore, EP4 receptor agonists have been suggested to have therapeutic potential as bronchodilators [58].
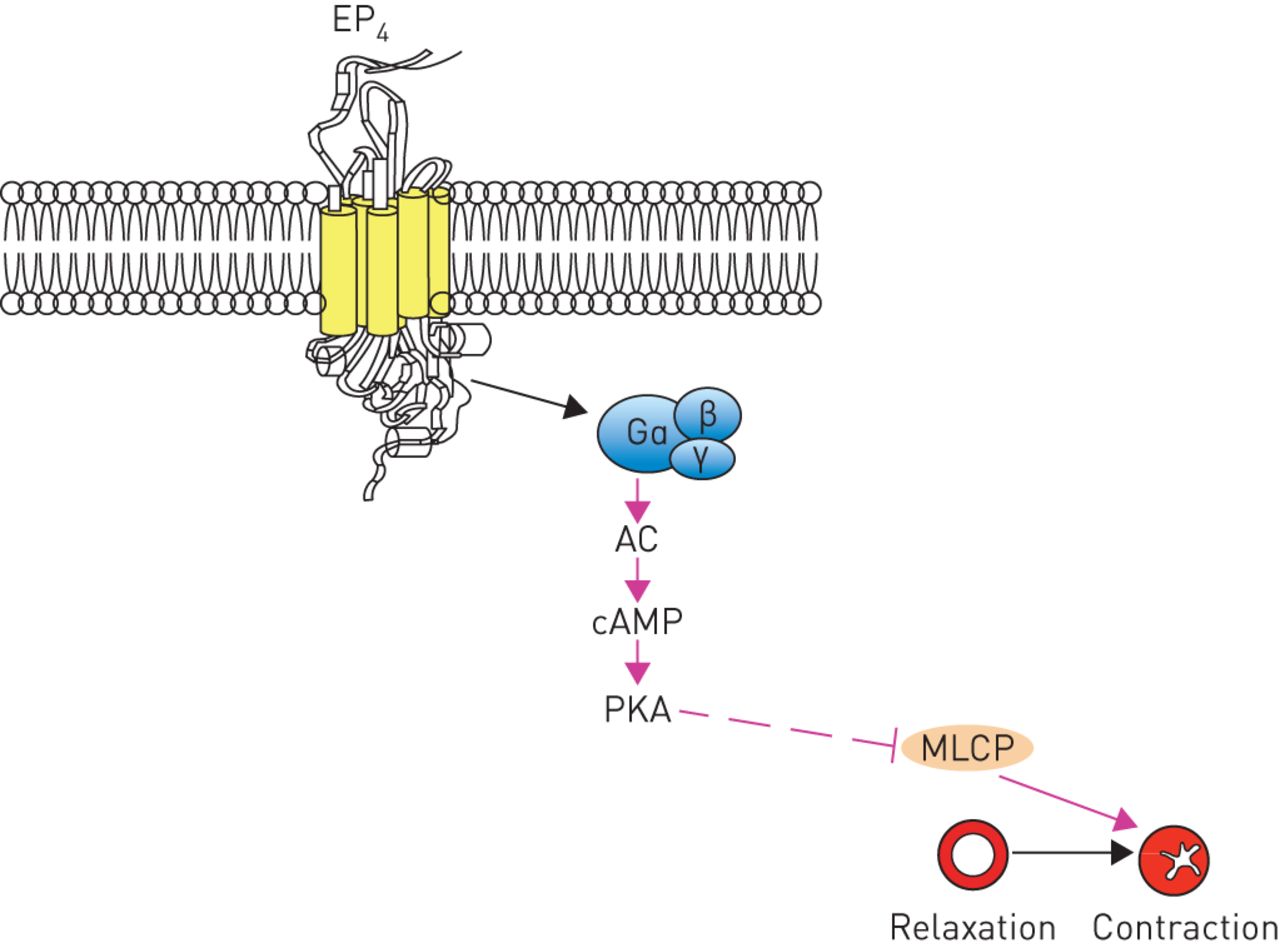
Activation of E-prostanoid receptor 4 (EP4) causes airway relaxation. AC: adenylyl cyclase; cAMP: cyclic adenosine monophosphate; PKA: protein kinase A; MLCP: myosin light chain phosphatase.
Some EP4 receptor agonists have been identified, including TCS2510, Compound 9, L-902688, ONO-AE1-734 and ONO-AE1-329. They have different binding affinity for the EP4 receptor, ranging from 4 nM (Compound 9) to 0.0097 μM (ONO-AE1-329). It has been shown that ONO-AE1-329 can relax human airways [56] and reverse histamine-induced contraction of ASM [59].
Interestingly, it has been that shown that activation of the EP4 receptor also elicits an anti-inflammatory effect in a range of disease-relevant models [58]. Furthermore, activation of EP4 is able to increase glucocorticoid receptor element-dependent transcription in human airway epithelial cells in a manner qualitatively similar to that reported with long-acting β-agonists [60].
These findings suggest a potential key role for EP4 receptor agonists in the treatment of airway disorders because a single molecule that is able to induce both anti-inflammatory and bronchodilator effects would be a preferred therapeutic option, especially if it could be developed as an oral medication, which is the best approach to increase patient's compliance [61]. Surprisingly, however, to date none of the available EP4 receptor agonists have been tested as bronchodilators in humans.
Rho kinase inhibitors
RhoA is a member of the Rho (Ras-homologous) family of small molecular guanosine triphosphatase related to Ras. It has been identified as the main upstream activator of Rho-associated coiled-coil-containing protein kinases (ROCK1 and ROCK2), which are serine/threonine kinases that can regulate the contraction of airways and other smooth muscle tissues, and is highly expressed in contractile ASM [62].
The RhoA/ROCK pathway plays an important role in the regulation of myosin light chain phosphatase (MLCP) (figure 1). Activation of ROCKs by RhoA phosphorylates the myosin-binding subunit that, when phosphorylated, inhibits the enzymatic activity of constitutively active MLCP, which regulates myosin light chain (MLC) phosphorylation in a Ca2+-independent manner. Inhibition of MLCP augments and sustains MLC phosphorylation, thereby promoting ASM contraction, which is termed Ca2+ sensitisation of smooth muscle contraction [63, 64].
Several studies in animal models of allergic bronchial asthma have shown an increased agonist-induced, RhoA-mediated contraction of ASM [65]. It has been demonstrated that inhibition of RhoA induces relaxation of methacholine-constricted human small airways [64]. Furthermore, repeated ROCK inhibition reduced the airway mechanical responses to antigenic challenge in an animal model of chronic airway inflammation [66]. These findings suggest that ROCK inhibitors may be effective bronchodilators.
Numerous ROCK inhibitors have been developed from a variety of distinct scaffolds and a number of them have been or are being evaluated in clinical trials, but only two have been approved for clinical use; fasudil for the treatment of cerebral vasospasm and ripasudil for the treatment of glaucoma in Japan and China [67]. There are, however, a number of novel ROCK inhibitors under development, including netarsudil (AR-13324), AMA0076 and Y-39983, but to date there have been no reported clinical studies with these drugs in patients with asthma or COPD.
The lack of studies with ROCK inhibitors is surprising because it has been suggested that these agents may act synergistically with other classes of bronchodilators, although this remains to be investigated [68]. Furthermore, ROCK inhibitors in conjunction with anti-IL-17 treatment are able to modulate airway responsiveness in mice with chronic allergic lung inflammation [69].
In any case, because of the abundant expression of these enzymes, mainly in the cardiovascular system, unwanted adverse effects could limit the development of ROCK inhibitors. However, local delivery to the lung, for instance as dry powder or through nebulisation, could limit the extent of these effects [70].
Calcilytics
The G-protein (heterotrimeric guanine nucleotide-binding protein)-coupled calcium-sensing receptor (CaSR) has recently emerged as a possible therapeutic target for asthma [71], although there is still limited information on the distribution and/or effects of CaSR in the lung. CaSR serves as the sensor for extracellular Ca2+ ([Ca2+]o) in the homeostatic serum Ca2+ feedback loop via the regulation of parathyroid hormone secretion [72]. Its activation causes the Gα11-dependent activation of PLC-β and the production of diacylglycerol (DAG) and IP3 from membrane-bound phosphatidylinositol 4,5-bisphosphate (figure 1). The increase in intracellular IP3 levels facilitates the release of Ca2+ from intracellular stores, whereas DAG activates protein kinase C and the p38 mitogen-activated protein kinase (MAPK) pathway [73].
Functional CaSRs are present in adult human ASM and regulate basal and agonist-induced [Ca2+]i, as well as contractility [74]. CaSRs are upregulated and constitutively activated in airways obtained from asthmatic patients [75]. It has been documented that the sputum from asthmatic patients contains elevated levels of polycationic peptides that are known to activate the CaSR and their effects on ASM are blocked by calcilytics [74].
Calcilytics, which are negative allosteric modulators of the CaSR, have been developed as drugs to affect Ca2+ levels systemically and mouse model studies indicate that they are able to rectify the hypocalcaemia and hypercalciuria associated with autosomal dominant hypocalcaemia [74]. Furthermore, it has been documented that their pulmonary delivery can suppress airway hyperresponsiveness in mouse asthma models, having no changes in blood levels of Ca2+ because administration directly to the lung avoids systemic exposure [74]. They also inhibit airway inflammation in both short- and long-term murine asthma models to a degree at least comparable to that of inhaled corticosteroids [75].
These findings suggest a potential role for a novel bronchodilator strategy. In effect, the use of inhaled calcilytics is under evaluation in preclinical models [74]. The novel calcilytic B2.1-E1 suppressed CaSR in vitro, and elicited parathyroid hormone release in vivo [76]. In addition, B2.1-E1 suppressed airway hyperresponsiveness and inflammation in mice. The challenges for the pharmaceutical industry are to design compounds that select conformations of the CaSR that preferentially target a particular signalling pathway, providing an approach for modulating CaSR function in a tissue- and disease-specific manner [77].
Agonists of PPAR-γ
PPAR-γ, which is a regulator of lipid and glucose metabolism and is the target for the thiazolidinedione class of synthetic antidiabetic agents such as pioglitazone, rosiglitazone, troglitazone, and ciglitazone, is widely expressed in the lung [78]. In particular, increased expression levels of PPAR-γ in ASM, epithelium and mucosal eosinophils and macrophages have been found in human airway biopsies from patients with asthma [78]. When activated, PPAR-γ exhibits several biological functions that are performed via regulation of transcription factors, notably stimulating the nuclear factor-κB-negative regulator IkBα [79].
Some preclinical studies have documented that PPAR-γ ligands are also able to relax ASM in vitro including the small airways [80, 81], particularly under conditions of impaired or limited β2-adrenoceptor responsiveness [81] but the potential roles of PPAR-γ agonists as bronchodilators have been poorly characterised. In trachea from naïve mice and guinea pigs and trachea and precision-cut lung slices from a mouse model of chronic allergic airways disease, rosiglitazone was less potent than β2-adrenoceptor agonists [82]. Its effects were additive with salbutamol and isoprenaline and only rosiglitazone maintained potency and full efficacy in maximally contracted airways or after allergen challenge [82]. Rosiglitazone was able to reverse salbutamol-induced β2-adrenoceptor tolerance in both tracheal muscular tissues derived from homologously desensitised guinea pigs and human ASM cells with a molecular mechanism that must still be clarified [83]. Relaxation to rosiglitazone was ascribed to accumulation of the dilator prostanoid PGE2 through inhibition of its breakdown [80].
Nonetheless, the efficacy of PPAR-γ agonists as asthma drugs continues to be controversial. Rosiglitazone induced a small improvement in lung function in patients with asthma [84], whereas pioglitazone did not result in any improvements [85].
Agonists of the relaxin receptor 1
Relaxin-2 belongs to the relaxin peptide family, a family of seven peptide hormones (three relaxin peptides, relaxin-1, -2 and -3 and four insulin-like peptides 3, 4, 5 and 6) involved in the activation of GPCR [86]. It is the major stored and circulating form of relaxin. Relaxin-2 consists of two chains (A and B) that are cross-linked by two inter-chain disulfide bonds and one intra-A-chain disulfide bond [87]. The B chain contains two arginine residues located on the same surface of the α-helix at positions 13 and 17. These residues are required for the binding of relaxin to its cognate GPCR, relaxin family peptide receptor 1 (RXFP1), which is a leucine-rich repeat containing GPCR [87]. RXFP1 is expressed in both airway epithelial cells and smooth muscle [88].
A potential benefit of pharmacologically modulating the relaxin-2–RXFP1 axis in lungs has been demonstrated using pure porcine relaxin in guinea pigs sensitised with ovalbumin [89]. This treatment counteracted the pulmonary asthma-like response to the inhaled antigen. Further information has been produced using the investigational drug serelaxin, the recombinant human form of relaxin-2 that combines the effects as a bronchodilator and antifibrotic agent [86].
Serelaxin has been demonstrated to elicit relaxation in airways and precision-cut lung slices from mice and guinea pigs via epithelial-dependent and epithelial-independent mechanisms, likely via RXFP1 activation and generation of nitric oxide (NO), prostaglandins and cAMP/cGMP [88]. In rat trachea precontracted with methacholine, serelaxin caused a partial relaxation relative to isoprenaline, although it had a slower onset of action. However, the onset of relaxation was improved when serelaxin was combined with rosiglitazone. Serelaxin also increased the salbutamol-induced relaxation of large and small human airways [90]. The signalling pathways, whereby serelaxin elicits relaxation of ASM and increases the potency of salbutamol, seem to be independent of cAMP-dependent pathways downstream of β2-adrenoceptor signalling and may, at least in part, rely on the generation of epithelial-derived relaxing factors [86].
Regrettably, the half-life of relaxin-2 in vivo is ∼10−20 min [91], which suggests that the mode of treatment for serelaxin is continuous infusion for 48 h. In order to overcome this issue, several approaches have been suggested but not yet evaluated in the clinical setting. The development of B-chain only agonists for RXFP1 has been considered. B7-33 was shown to bind to RXFP1 and preferentially activate the extracellular signal-regulated kinase (ERK) pathway over cAMP in cells that endogenously expressed RXFP1 (figure 1) [92]. In mouse models with ovalbumin-induced chronic allergic airways disease, B7-33 significantly diminished airway epithelial thickening, amount of total lung collagen (a benchmark of fibrosis), and airway hyperresponsiveness (a benchmark of lung dysfunction) to a similar magnitude to native relaxin-2 [93]. An alternative approach is to target RXFP1 using small molecules that act as either agonists or alternatively augment the agonist activity of relaxin-2 as positive allosteric modulators [86]. ML290 was the most promising of these small molecules and, consequently, novel ML290 analogues have been generated [94].
Recently, a semisynthetic methodology for the synthesis, through the cysteine introduced in recombinant relaxin, generated a series of potent fatty-acid-conjugated relaxin-2 analogues that have longer half lives in vivo when evaluated in rodents [95]. The resultant novel relaxin analogue, R9-13, represents the first long-acting relaxin-2 analogue.
Soluble guanylyl cyclase activators
NO donor compounds are another class of drugs that have been shown to relax human airways in vitro raising the possibility that such compounds may prove to be useful as novel bronchodilators [5]. Nanomolar concentrations of NO bind to the reduced Fe2+ (ferrous) heme moieties on the heterodimer soluble guanylyl cyclase (sGC), a eukaryotic NO receptor consisting of an α-subunit and a β-subunit, which in turn converts guanosine triphosphate to cGMP that subsequently induces protein kinase G phosphorylation, and changes in activity of other effector proteins, including PDEs, ion channels and ion pumps, all leading to ASM relaxation [96]. However, inflammation has been shown to desensitise sGC toward its natural activator NO, leading to a diminished bronchodilation [97]. In effect, there is documentation that expression of sGC is decreased in smokers and COPD patients [98], and also in human ASM cells derived from asthma [98].
The possibility that NO donor compounds may prove to be useful as novel bronchodilators is not solid and this is not only because it is problematic to carefully target NO release to lungs at a concentration capable of inducing beneficial action and avoiding the onset of possible adverse effects, particularly on the cardiovascular system [3], but also because of the potential lack of response, the development of tolerance and the nonspecific interactions of NO with biomolecules (such as superoxide, leading to formation of peroxynitrite) [96]. For these reasons, it has been suggested that greater benefits could be obtained by activating the NO–sGC–cGMP pathway that bypasses the need for NO and instead activates sGC directly, causing bronchodilation, and/or inhibiting the NO-induced formation of proinflammatory molecules [96].
The sGC stimulator cinaciguat (BAY 41-2272) and activator BAY 60-2770 reversed airway hyperresponsiveness in mice with allergic airways inflammation and were able to restore lung function [99], whereas the pharmacologic activation of sGC by BAY 58-2667 attenuated bronchial hyperresponsiveness in cigarette smoke-exposed mice [97]. Furthermore, activation of sGC induced significant bronchodilation in human lung slices, in some cases to an extent similar to that observed with isoprenaline or formoterol [100]. In contrast to these studies, no changes in lung functional parameters were observed in guinea pigs chronically exposed to cigarette smoke, although sGC stimulation exerted beneficial effects on lung parenchyma and pulmonary vasculature [101].
Pepducins
Pepducins are cell-penetrating lipidated peptides incorporating sequences from GPCRs [102]. They cross the plasma membrane and anchor to the cytosolic interface to activate or inhibit the signalling of receptors. Therefore, they have potential as therapeutic agents [103].
GPCRs are rapidly uncoupled from G-proteins resulting in signal desensitisation not only by phosphorylation by GPCR kinase (GRK), but also by β-arrestin binding that increases when GRK phosphorylates the GPCR [104]. It seems likely that a G-protein-biased ligand unable to induce β-arrestin recruitment may cause less tachyphylaxis and would be a more efficient therapeutic agent [105].
It is now recognised that β-arrestins are also capable of triggering signalling cascades independent of G-protein signalling and β2-adrenoceptors can signal not only via activation of G-proteins, but also via β-arrestins [105]. Some manifestations of the signalling capacities of β-arrestins have also been appreciated and, in effect, numerous substrates including ERK, cJun N-terminal kinase, p38 MAPK, phosphoinositide-3-kinase, Akt, and RhoA have been identified for β-arrestin-dependent signalling in the cytoplasm [106] Therefore, interfering with β-arrestin-driven processes could offer novel strategies for therapeutic intervention [107]. Conveniently, ligands could be biased or “functionally selective” toward either a G-protein or β-arrestin-mediated pathway (figure 4) [107, 108]. Agonists that show a higher potency to specific signalling pathways over others are known as “biased agonists” and have a better therapeutic index [109].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Biased various G-protein coupled receptor (GPCR) signalling. Biased GPCR ligands are able to engage with their target receptors in a manner that preferentially activates only G-protein- or β-arrestin-mediated downstream signalling. GRK: GPCR kinase.
Pepducins that target specific intracellular loops of GPCRs have recently emerged as effective allosteric modulators of GPCR activity [110]. Pepducins from the third intracellular loop (ICL)3) of the β2-adrenoceptor could directly activate Gs in the presence or absence of receptor, while β-arrestin-biased pharmacology was demonstrated mostly with pepducins derived from ICL1 sequences [111]. The current experimental evidence suggests that direct targeting of G-proteins, or manipulation of GRK and arrestin function, can promote bronchodilation [103]. The pepducin ICL3–9 derived from ICL3 of β2-adrenoceptor showed Gs-biased signalling, limiting β-arrestin-mediated desensitisation and potential tachyphylaxis from the chronic use of β2-adrenoceptor agonists [112]. AT1-2341 is a pepducin derived from the ICL1 that promotes specific Gi-mediated signalling without G13-coupling or β-arrestin recruitment [113].
In any case, lipidated peptide agonists and antagonists of other classes of cell-surface receptors have also been developed [110]. Pepducins that function as broad-based antagonists of Gq signalling have been designed [104]. The M3 mAChR, cysteinyl leukotriene, H1 histamine, thromboxane, endothelin-A/B and others are Gq-coupled receptors that are activated in asthma or COPD. P4pal-10 is a protease activated receptor 4-derived pepducin that exhibits efficacy toward multiple Gq-coupled receptors [113].
Conclusion
Considering the pivotal importance of bronchodilation in the pharmacological treatment of patients with asthma or COPD [1] and also the safety concerns of currently used inhaled drugs, the potential for a loss in bronchoprotection of the β2-adrenoceptor agonist class, and lack of disease-modifying properties of inhaled bronchodilators [7], there is a real interest within researchers and also the pharmaceutical industry in developing novel bronchodilators.
As we have previously pointed out, there are several new opportunities. However, they are mostly in a preclinical phase. We still do not know which classes will actually be developed for clinical use once it has been established that the possible clinical benefit will outweigh the impact of any adverse effect. However, regardless of this, it will be necessary to establish whether these new classes will be able to replace, if necessary, the classes currently available, because the extent of bronchodilation that they will elicit will be higher or, at least equal to that of existing bronchodilators, with a greater safety profile. In any case, it is also possible that these new classes may be a useful addition, rather than a substitution, of the bronchodilator therapy currently used, in order to achieve further optimisation of bronchodilation.
Footnotes
Submitted article, peer reviewed
Conflict of interest: M. Cazzola has nothing to disclose.
Conflict of interest: P. Rogliani has nothing to disclose.
Conflict of interest: M.G. Matera has nothing to disclose.
- Received July 30, 2019.
- Accepted September 3, 2019.
- Copyright ©ERS 2019.
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










