





Abstract
Severe steroid-resistant asthma is clinically important, as patients with this form of the disease do not respond to mainstay corticosteroid therapies. The heterogeneity of this form of asthma and poor understanding of the pathological mechanisms involved hinder the identification of therapeutic targets and the development of more effective therapies. A major limiting factor in the understanding of severe steroid-resistant asthma is the existence of multiple endotypes represented by different immunological and inflammatory phenotypes, particularly in adults. Several clinical and experimental studies have revealed associations between specific respiratory infections and steroid-resistant asthma in adults. Here, we discuss recent findings from other authors as well as our own studies that have developed novel experimental models for interrogating the association between respiratory infections and severe steroid-resistant asthma. These models have enabled the identification of new therapies using macrolides, as well as several novel disease mechanisms, including the microRNA-21/phosphoinositide 3-kinase/histone deacetylase 2 axis and NLRP3 inflammasomes, and highlight the potential of these mechanisms as therapeutic targets.
Abstract
Severe steroid-resistant asthma is a significant clinical problem and recent advances in understanding the mechanisms of pathogenesis enable the identification of novel therapeutic approaches http://bit.ly/2mQpF3n
Introduction
The Global Initiative for Asthma (GINA) currently defines asthma as a heterogeneous disease that is characterised by inflammation of the airways and a clinical history of wheezing, cough, tightness of chest and shortness of breath varying with time and intensity, as well as expiratory airflow limitation [1]. Asthma is classically considered as a disease driven by increased type 2 immune responses and eosinophilic inflammation [2–5]. However, recent evidence shows that asthma can be associated with elevated type 1/17 immune responses that are associated with non-eosinophilic (i.e. neutrophilic) inflammation and tend to be present in adult patients with moderate to severe asthma [2, 6]. Importantly, gene–environmental interactions play a vital role in the progression of the disease, with specific insults to the respiratory epithelium [7] inducing the release of pro-inflammatory mediators that recruit inflammatory cells to the airways. Exogenous factors such as respiratory viral and bacterial infections, a high-fat diet and/or obesity, air pollution and cigarette smoke exposure have all been linked with severe forms of asthma in adults as well as with exacerbations of the disease [2, 8–20]. Here, we review the mechanisms of pathogenesis of severe steroid-resistant (SSR) asthma in adults. Children can also have severe asthma, which may have different pathomechanisms; these have been reviewed recently elsewhere [21–23].
Anti-inflammatory corticosteroids are the mainstay therapy for asthma and are often administered in conjunction with short- or long-acting β2-adrenoceptor agonists (bronchodilators) [2, 24]. These combination therapies allow most asthmatics to gain control of their disease symptoms by modulating type 2 cytokine responses and accompanying inflammation, although they have broad-spectrum, nonspecific anti-inflammatory activity. Importantly, however, these therapies are not cures, and long-term, high-dose corticosteroid therapy is associated with many side-effects [4, 24, 25]. Critically, 5–10% of asthmatics do not respond to steroid-based therapies, are more likely to have severe disease and, due to the paucity of effective treatment options, disproportionately account for 50–80% of all asthma-associated healthcare costs [26, 27]. SSR asthma is strongly associated with non-eosinophilic endotypes of the disease, including neutrophilic asthma, and often involves activation of innate immune responses, particularly those mediated by Toll-like receptor (TLR)2 and TLR4 responses, and NLRP3 (nucleotide-binding oligomerisation domain-like receptor family, pyrin domain-containing 3) inflammasome/interleukin (IL)-1β responses [28–32]. Schwartz et al. [33] first described steroid-resistant asthma in a small asthmatic subgroup with poor disease control that did not respond to high-dose oral steroid therapy. With progressive refinement of diagnostic criteria, SSR asthma in adults is now defined as ineffectiveness in achieving >15% improvement in forced expiratory volume in 1 s after 14 days of oral steroid treatment [27, 34]. SSR asthma is the major clinical issue in asthma management and there is an urgent need for therapies that more effectively suppress the symptoms of disease (figure 1).
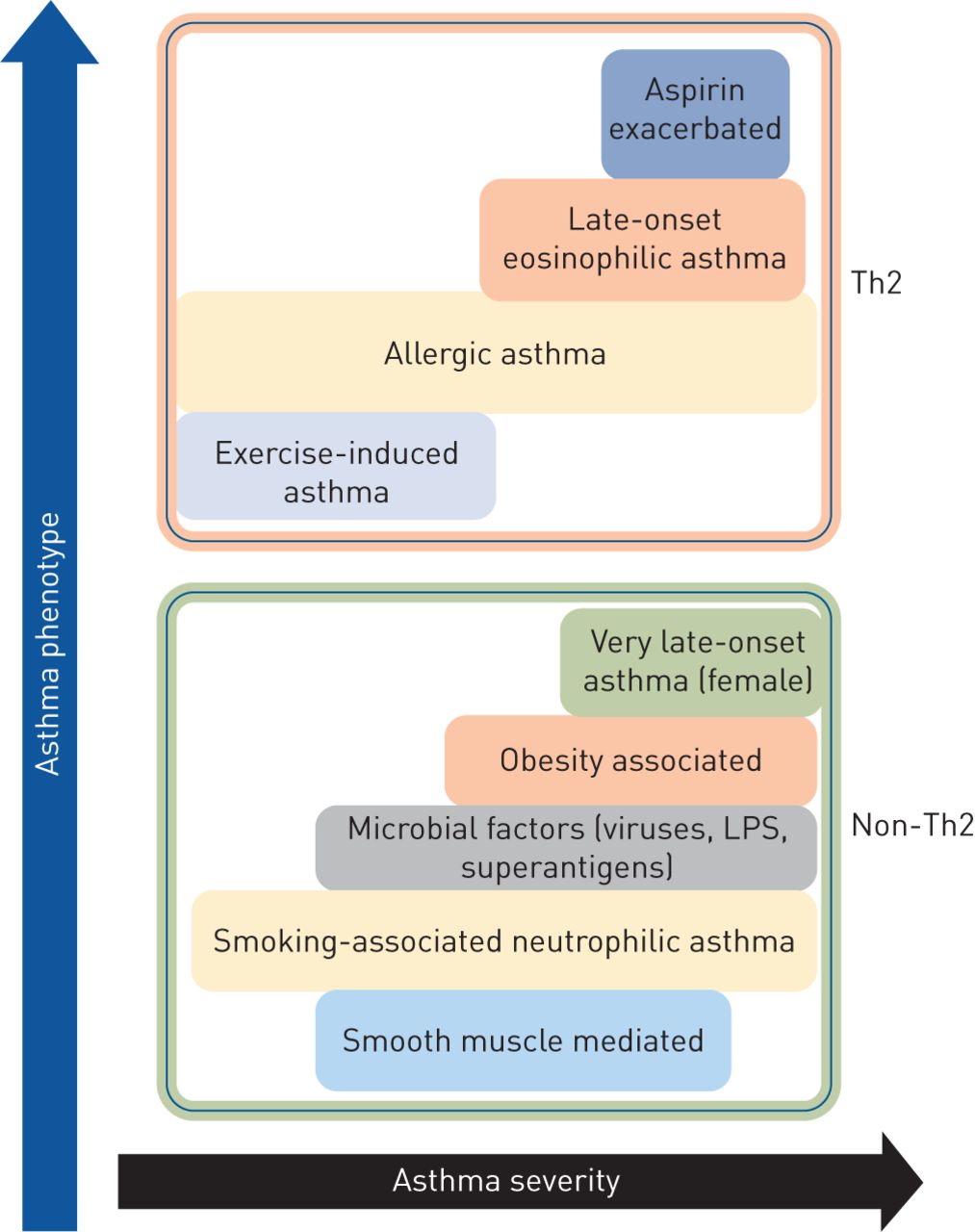
Classification of asthma phenotypes and disease severity based on T-helper cell type 2 (Th2) and non-Th2 characteristics. LPS: lipopolysaccharide.
Mechanisms of steroid resistance
Glucocorticoids (a class of anti-inflammatory corticosteroid) are the mainstay therapy for asthma and are administered through inhaled or oral formulations. Inhaled corticosteroids (ICSs) are synthetic, lipophilic glucocorticoids that, upon inhalation, readily diffuse into the airway tissue and bind and activate cytosolic glucocorticoid receptors (GRs; also called nuclear receptor subfamily 3, group C, member 1 (NR3C1)) [35, 36]. The anti-inflammatory effects are mediated by the GRα isoform via ligand-dependent transcription factors, while GRβ acts as a dominant inhibitor of GRα [37]. GRα homodimers are actively transported into the nucleus by importin-α and importin-13 and bind to a glucocorticoid response element (GRE) to regulate gene transcription [25, 36, 38]. The interaction with transcriptional co-activators, including CBP (cAMP response element binding (CREB)-binding protein), leads to the acetylation of core histones associated with anti-inflammatory glucocorticoid response genes and induction of RNA polymerase II-mediated gene transcription. This phenomenon is known as transactivation. Alternatively, activated GR interacts with CBP proteins complexed with promoter regions of pro-inflammatory genes, such as NF-κB and activator protein (AP)-1, causing suppression of pro-inflammatory gene transcription via inhibition of histone acetylation and preventing DNA access to RNA polymerase II [39–41]. This process is called transrepression.
A large body of clinical and experimental evidence shows that defective GRα expression and activity impairs the anti-inflammatory effects of glucocorticoids and plays important roles in the induction of steroid resistance. Interestingly, other studies show that the higher levels of IL-2, IL-4 and IL-13 gene expression result in local cytokine secretion and steroid resistance, which further alters GRα translocation [34, 42]. A study by Goleva et al. [43] reported that specific silencing of GRβ in bronchoalveolar lavage macrophages of individuals with SSR asthma resulted in improved GRα activity. In another study, Irusen et al. [44] reported that activation of p38 mitogen-activated protein kinase results in the phosphorylation and impaired function of GRs. Moreover, defective nuclear translocation of GRs results in reduced interaction with GREs and GR:GRE binding is affected by increased NF-κB, AP-1, c-Fos and c-Jun N-terminal kinase responses [34, 42, 45, 46]. Defective nuclear translocation of GRs reduces expression and activity of histone deacetylase (HDAC)2 in patients [47]. Importantly, many patients with SSR asthma have normal nuclear translocation of GR but reduced GR:GRE binding affinity [48, 49], suggesting that SSR asthma can also be induced by dysfunction of a different mechanism. Interestingly, reduced HDAC2 expression and activity is associated with steroid insensitivity and more severe disease in both asthma and COPD [50–52], suggesting that deficiencies in transcriptional co-repressor expression and activity may be important in the pathogenesis of SSR asthma.
Role of respiratory infections in SSR asthma
SSR asthma is a serious clinical and socioeconomic problem that is associated with lung function impairment, airway hyperresponsiveness (AHR) and more intense disease exacerbations leading to hospitalisation. Limited treatments are available, including corticosteroid therapy, but none are cures or broadly effective. A crucial drawback is that there are several subsets of SSR asthma with different immunological and inflammatory phenotypes that are likely to drive steroid resistance through different mechanisms [2, 5, 6, 53]. Increasing clinical and experimental evidence strongly implicates specific respiratory infections in the pathogenesis of SSR asthma. We have developed novel and unique animal models of respiratory infection-induced SSR asthma and used them to demonstrate that Chlamydia, Haemophilus influenzae, influenza A virus (IAV) and respiratory syncytial virus (RSV) respiratory infections induce steroid resistance of the key disease features [16–18, 30, 54–56].
We first established Chlamydia, Haemophilus, IAV and RSV respiratory infections and allergic airway disease (AAD) in BALB/c mice separately. Clinical studies show that infections drive more severe disease in established disease [30, 55, 56]. Thus, AAD was first established and then infection was induced. Mice were systemically sensitised with ovalbumin (Ova) and aluminium hydroxide intraperitoneally, followed 10–12 days later by inhaled Ova challenge to induce AAD. Other mouse models of allergic asthma widely use other allergens, such as house dust mite [57–59], Alternaria [60] or cockroach [61] antigen. Our mice were left for 20 days and were re-challenged on days 33–34 of the model, in order to model an allergen-induced exacerbation of disease. To induce severe steroid-resistant AAD (SSRAAD), we induced infections in allergic mice after AAD was initially established, to model the human scenario. Infections were resolved before allergen-induced exacerbation of AAD. To model ICS treatment and examine the response to steroid treatment, mice were administered the steroid dexamethasone (DEX) for the final 3 days of the protocol. Key disease features were assessed 1 day after the final Ova challenge, to assess the impact of resolved infection on the disease phenotype. Importantly, we found that infection drives a severe form of AAD that does not respond to steroid treatment. These protocols have resulted in novel experimental models of Chlamydia-, Haemophilus-, IAV- and RSV-induced SSR asthma that each represents unique endotypes of SSR asthma in humans (table 1).
Role of infections in the pathogenesis of severe steroid-resistant asthma and their response to steroid therapy
Chlamydia respiratory tract infection in asthma corresponds to increased airway neutrophils, which predicts the presence of this pathogen and of steroid resistance [55]. It has also been linked to severe asthma in children [62–64]. Chlamydia respiratory infection in mice with AAD results in increased type 1- and 17-associated immune responses, neutrophilic inflammation, suppression of type 2 and eosinophilic inflammatory responses [18, 30, 55, 56], and steroid-resistant inflammation and AHR. In early life, Chlamydia respiratory infections promote more severe experimental asthma in mice by switching type 1 phenotype and inducing IL-13 and TRAIL (tumour necrosis factor-related apoptosis-inducing ligand) responses [17, 65–67]. These models of early-life infection-induced severe asthma are also steroid resistant (P.M. Hansbro, unpublished data).
H. influenzae is frequently isolated from the airways of SSR asthma patients and correlates with oxidative stress markers, neutrophilic inflammation, more severe airflow obstruction and steroid resistance [19, 20]. Experimentally, H. influenzae respiratory infection drives type 17-associated immune responses, airway neutrophilia, steroid resistance and impaired macrophage phagocytosis [16, 27, 54–56].
Viral respiratory tract infections are strongly associated with the induction of asthma in early life and in exacerbations of asthma that are resistant to mainstay therapies. IAV and RSV infections in murine models of asthma result in exaggerated airway eosinophil influx and type 1 and 2 immune responses, mixed eosinophilic and neutrophilic airway inflammation, and steroid resistance of airway inflammation and AHR [56, 68, 69].
Thus, these mouse models are highly representative of the different immune and inflammatory phenotypes of SSR asthma in humans but in each case the inflammation and AHR are steroid resistant. We have interrogated these models to identify critical roles for microRNA (miR)-21/phosphatase and tensin homologue (PTEN)/phosphoinositide 3-kinase (PI3K)/HDAC2 and NLRP3 inflammasome/IL-1β responses in the pathogenesis of experimental and clinical SSR asthma [27, 55, 56]. Importantly, the infections can activate innate immune responses and induce neutrophilic inflammation and type 1 and 17 immune responses that are also associated with SSR asthma [2]. We have identified potential new treatments using macrolides, miR-21-specific antagomirs, and PI3K and NLRP3 inflammasome inhibitors [30, 55, 56]. Our models can be interrogated to identify potentially important relationships between microbiome load in pulmonary and extrapulmonary compartments and key disease features. Such analyses should be done rigorously to identify novel disease-causing factors.
Novel treatment strategies for SSR asthma
Macrolides
Macrolide antibiotics are a first-line treatment for bacterial infections and exert their effects by blocking bacterial protein synthesis, adherence, motility and biofilm formation [70, 71]. Importantly, recent clinical and experimental evidence shows that they also have anti-inflammatory properties. Macrolides have been shown to be immunomodulatory in severe asthma and can enhance the steroid response by suppressing inflammatory cell infiltration and AHR [72].
We recently used our novel mouse models of Chlamydia and Haemophilus respiratory infection-induced SSR asthma to examine the effects of treatment on key disease features with a macrolide (clarithromycin) or standard beta-lactam antibiotic (amoxicillin, which has no known anti-inflammatory properties) in the presence or absence of DEX [55]. Treatment with clarithromycin and amoxicillin had similar antimicrobial effects; however, only clarithromycin suppressed steroid-resistant airway inflammation and AHR in both eosinophilic and neutrophilic AAD. Importantly, clarithromycin exerted its beneficial effects by reducing type 2, tumour necrosis factor (TNF)-α and IL-17 responses. Treatment with amoxicillin had limited effects on key disease features but restored steroid sensitivity, probably by reducing bacterial load. By comparison, the effects of clarithromycin on suppressing features of AAD were independent of its antimicrobial effects. These data show that macrolide-based therapies may be effective in SSR asthma irrespective of the presence of respiratory infection.
A different study showed that treatment with the macrolide roxithromycin significantly improved lung function within 6 weeks in asthmatics with evidence of Chlamydia infection [73]. Similarly, in asthmatic children with evidence of Chlamydia infection, clarithromycin treatment reduced the risk and duration of wheezing [74]. Clarithromycin treatment of asthmatics with evidence of Chlamydia infection also reduced IL-5 levels in the airways [75].
In our study, we showed that treatment with clarithromycin reduced IL-17 responses and suppressed inflammation and AHR in both SSR and steroid-sensitive AAD [54, 55]. Treatment also reduced airway neutrophil count and the levels of sputum IL-8, neutrophil elastase and matrix metalloproteinase-9 [76]. These data demonstrate that clarithromycin treatment has antimicrobial as well as anti-inflammatory effects that suppress pro-inflammatory responses in different asthma endotypes with evidence of steroid-resistant disease features, and highlight its potential application in treating infection-induced SSR asthma.
The immunomodulatory effects of macrolides were elegantly demonstrated in a clinical study (AMAZES study) performed by Gibson et al. [77], who performed a randomised, double-blind, placebo-controlled parallel group trial to assess whether the macrolide azithromycin (500 mg, orally administered three times per week for up to 48 weeks) reduced asthma exacerbations in adults with symptomatic asthma. In this study, azithromycin was used as an add-on therapy to 1) ICS and long-acting β2-adrenoceptor agonist (n=345; 82%), 2) long-acting β2-adrenoceptor agonist with long-acting muscarinic antagonist (n=68; 16%), 3) long-acting muscarinic antagonist (n=5; 1.2%), or 4) theophylline (n=2; <1%). A total of 420 randomised asthma patients were assigned, 213 for an azithromycin group consisting of 144 (43%) patients with an eosinophilic endotype and 187 (57%) patients with a neutrophilic endotype, while 207 were treated with placebo control. Azithromycin reduced exacerbations down to 1.07 per patient-year compared to placebo with 1.86 per patient-year. Towards the end of the treatment regimen it was found through sputum culture that 37 patients (17 in the treatment and 20 in the placebo group) had detectable levels of potentially pathogenic microbes. Azithromycin significantly improved quality of life in asthma patients; however, its use was associated with diarrhoea. Nevertheless, these data show that the immunomodulatory effects of azithromycin may be an effective add-on therapy in severe asthma.
miR-21/PI3K/HDAC2 axis
The anti-inflammatory effects of corticosteroids are mediated by activation of GR/NR3C1 and deacetylation of histones by HDAC2, leading to suppression of pro-inflammatory gene transcription and/or induced expression of anti-inflammatory genes [36, 78]. Importantly, increased PI3K activity is known to decrease HDAC2 responses and diminish steroid responses [52]. Furthermore, increased PI3K activity induces type 17-associated immune responses and reduces interferon (IFN) responses, which allows entry and replication of pathogens in host cells, which can lead to further increases in PI3K activity [79, 80]. Several miRs have been implicated in the pathogenesis of asthma [81, 82] and, of these, miR-21 is known to be important in AAD pathogenesis. miR-21-deficient (miR-21−/−) mice with AAD have reduced eosinophilic inflammation and IL-4 levels, and elevated IFN-γ responses [81, 83]. Biological network-based transcriptome analysis of Ova-challenged miR-21−/− mice determined that dysregulation of IL-12/IFN-γ played an important role in the observed phenotype and implicated miR-21 as a key regulator of IFN-γ signalling and T-cell polarisation. Thus, the loss of miR-21 augments T-helper cell type 1 (Th1)-related delayed hypersensitivity [83]. Interestingly, miR-21 has also been shown to directly downregulate the expression of PTEN, which is an endogenous suppressor of PI3K activity [84]. A study by Kwak et al. [84] demonstrated that mice with Ova-induced AAD had reduced PTEN levels in the epithelial layer of bronchioles and that adenovirus-mediated overexpression of PTEN in AAD resulted in increased IL-4 and IL-5 levels in the airways and reduced AHR. Furthermore, intratracheal administration of pharmacological pan-PI3K inhibitors suppressed bronchial inflammation and AHR in mice with AAD, which highlights the potential for using PI3K inhibitors, such as wortmannin, in the treatment of severe asthma [85].
Recently, we examined the role of miR-21 in the pathogenesis of SSR asthma and assessed its potential for therapeutic targeting using our novel mouse models of Chlamydia, H. influenzae, influenza and RSV respiratory infection-induced SSR asthma [56]. We first profiled the expression of all known mouse miRs using microarray analysis and found that miR-21 was the most highly expressed in our mouse models. We confirmed this result by quantitative PCR and then showed that miR-21 expression in AAD is not reduced by steroid treatment. We measured the expression of several putative targets of miR-21 (identified by multiple target prediction algorithms) and found that PTEN was consistently downregulated in a miR-21-enriched environment and that this was associated with increased PI3K responses (elevated nuclear levels of phosphorylated AKT). Importantly, we showed that these effects were associated with reductions in both nuclear levels of HDAC2 and lung expression of NR3C1. This led us to hypothesise that a novel miR-21/PTEN/PI3K/HDAC2 pathway plays an important disease-causing role in SSRAAD (figure 2).
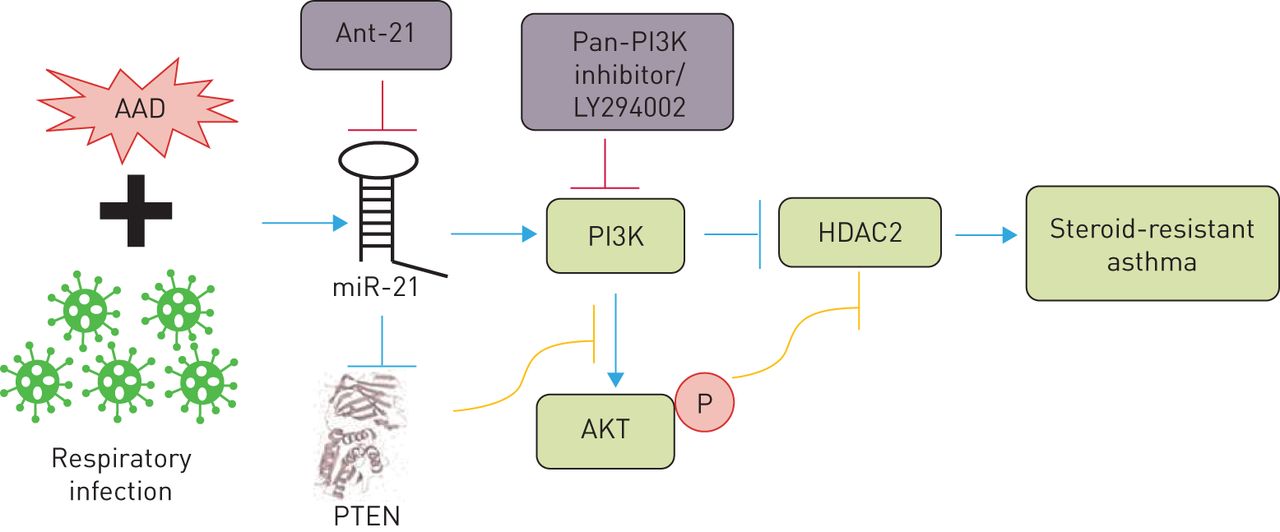
A microRNA (miR)-21/phosphoinositide 3-kinase (PI3K)/histone deacetylase (HDAC)2 axis drives severe steroid-resistant asthma and may be targeted using miR-21-specific inhibitors (such as antagomiR (Ant)-21) and/or PI3K inhibitors (such as LY294002). AAD: allergic airway disease; PTEN: phosphatase and tensin homologue.
We then demonstrated the functional role of this pathway and its potential for therapeutic targeting using our mouse models of infection-induced SSRAAD. Intranasal treatment with a miR-21 inhibitor, antagomiR-21, completely suppressed lung miR-21 expression, increased PTEN expression, reduced PI3K activity and increased nuclear levels of HDAC2. Importantly, treatment with antagomiR-21 effectively suppressed key disease features in SSRAAD. Treatment with antagomiR-21 alone had no effect on airway inflammation (i.e. total leukocytes, eosinophils, neutrophils, macrophages or lymphocytes) but restored sensitivity to steroid treatment in SSRAAD (numbers of all cells were reduced after DEX treatment). Interestingly, treatment with antagomiR-21 in the absence of steroids completely suppressed steroid-resistant AHR in SSRAAD. We also observed similar effects using the pan-PI3K inhibitor LY294002. Notably, all disease features were completely suppressed by antagomiR-21 or LY294002 back to baseline levels achieved with steroid treatment of steroid-sensitive AAD in the absence of infection. Furthermore, we observed similar effects in Haemophilus-, IAV- and RSV-induced SSRAAD.
These data are especially important because Chlamydia/Haemophilus respiratory infections are strongly implicated in type 1/17 immune response-associated, steroid-resistant, neutrophilic asthma, and IAV/RSV infections are strongly implicated in type 1-associated, steroid-resistant, eosinophilic asthma. Collectively, our data show that targeting miR-21 and/or PI3K is likely to be effective across several endotypes of SSR asthma (figure 2) [56].
NLRP3 inflammasome
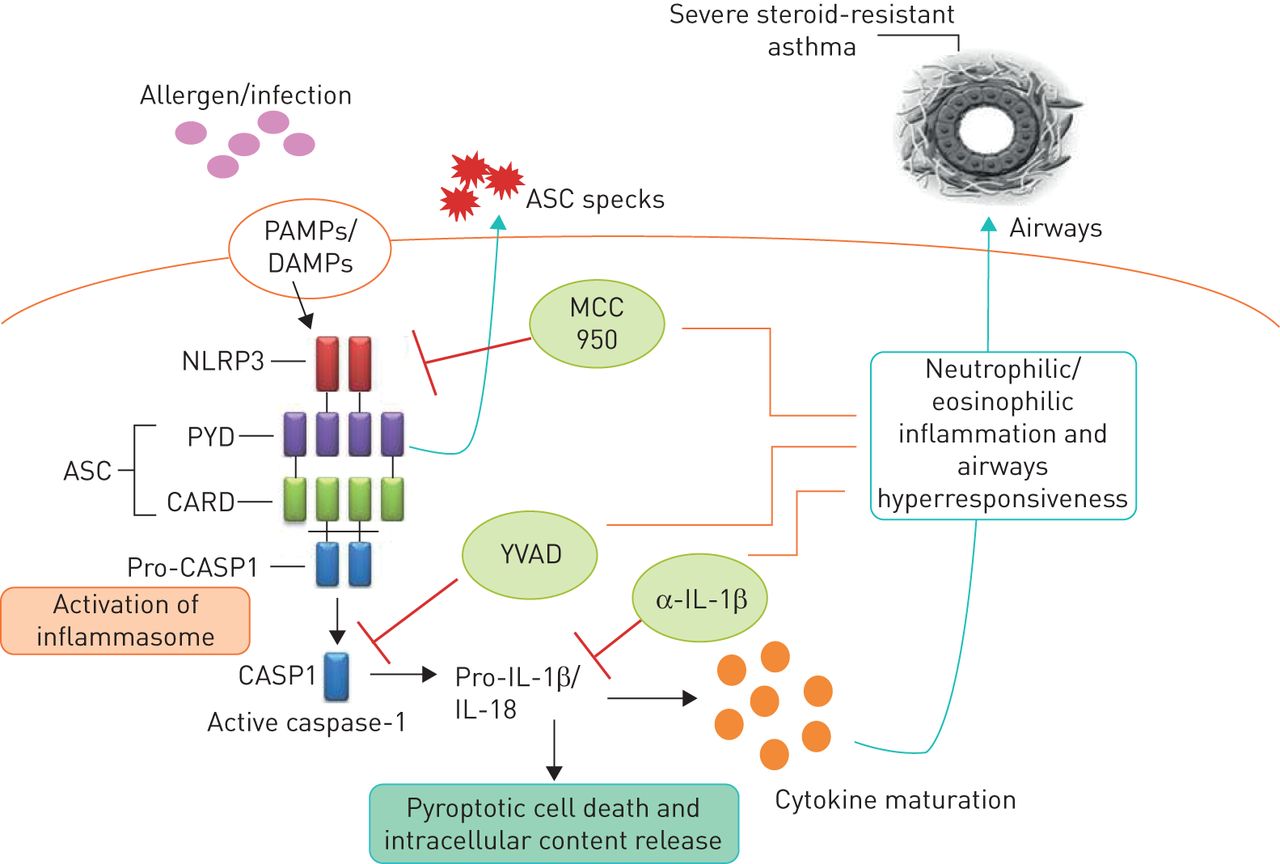
Inflammasomes are multiprotein signalling complexes that control the maturation and release of potent pro-inflammatory cytokines that are produced in response to endogenous, exogenous and pathogenic risk signals [28, 86]. The NLRP3 inflammasome is composed of an apoptosis-associated speck-like protein containing a pyrin domain, a caspase recruitment domain and a pro-caspase-1 domain [86]. The expression and assembly of inflammasomes occurs as a result of stimulation by microbe-derived pathogen-associated molecular patterns such as lipopolysaccharide and double-stranded DNA. A second signal in the form of damage-associated molecular patterns induces the activation of assembled inflammasomes that cleave and activate pro-caspase-1, which in turn cleaves pro-IL-1β and pro-IL-18 into active IL-1β and IL-18, respectively [28, 31, 86]. Thus, inflammasome activation is essential for caspase-1-dependent IL-1β processing, maturation and secretion (figure 3).

{kind=link}
{kind=link}
{kind=link}
Role of the NLRP3 inflammasome in severe steroid-resistant asthma. An infection and/or allergen promotes NLRP3 inflammasome-mediated cleavage of pro-caspase-1 into caspase-1 (CASP1), which then cleaves pro-interleukin (IL)-1β into active IL-1β, which drives steroid-resistant asthma. Neutralising anti (α)-IL-1β antibodies, a CASP1-specific inhibitor (YVAD) and a potent and highly specific NLRP3 inflammasome inhibitor (MCC950) may have therapeutic utility in severe steroid-resistant asthma. PAMPs: pathogen-associated molecular patterns; DAMPs: damage-associated molecular patterns; ASC: adaptor protein apoptosis-associated speck-like containing a caspase recruitment domain; PYD: pyrin domain; CARD: caspase recruitment domain.
Neutrophilic asthma is associated with increased levels of NLRP3 and caspase-1 in the airways, exaggerated IL-1β responses, and Th17 differentiation and IL-17 production that are all implicated in AHR and more severe forms of asthma [30, 31]. Simpson et al. [87] first demonstrated that neutrophilic asthma is associated with increased sputum expression of TLR2, TLR4, IL-1β and IL-8 and increased levels of lipopolysaccharide. These observations are supported by another clinical study by Baines et al. [88], which used sputum RNA expression profiling to show that genes in the IL-1 and TNF-α/NF-κB signalling pathways were overexpressed and correlated with key clinical parameters and neutrophilic airway inflammation in asthma. Notably, Simpson et al. [89] showed that sputum macrophages are a major source of the elevated NLRP3, caspase-1, caspase-4, caspase-5 and IL-1β levels they observed. Furthermore, Darville et al. [90] observed that ATP-mediated P2X7R signalling is essential for immunity against Chlamydia, and Chlamydia, Haemophilus, IAV and RSV infections all induce NLRP3 inflammasome-induced caspase-1-mediated activation and release of IL-1β [91–94].
We recently showed that NLRP3, caspase-1 and IL-1β responses are increased in the lung in Chlamydia infection-induced experimental SSR asthma [30] and that sputum NLRP3 and IL-1β expression correlate with key clinical parameters of human SSR asthma, including increased percentage and absolute numbers of neutrophils in the airways, loss of asthma control and reduced lung function [30]. We then interrogated the functional roles and potential for therapeutic targeting of NLRP3, caspase-1 and IL-1β in experimental SSR asthma by intranasally administering MCC950 (a potent and highly selective NLRP3 inflammasome inhibitor [95]), ac-YVAD-cho (a selective caspase-1 inhibitor) or neutralising anti-IL-1β monoclonal antibody during Chlamydia and Haemophilus infection-induced SSRAAD. Increased lung IL-1β response was suppressed by all three treatments and this effect was associated with the suppression of steroid-resistant neutrophilic airway inflammation and AHR. We then demonstrated that the disease-causing effects of infection are driven by IL-1β and neutrophilic inflammation by recapitulating the key features of neutrophilic SSRAAD in mice with eosinophilic, steroid-sensitive AAD using recombinant mouse IL-1β and anti-Ly6G (blocks recruitment of neutrophils). We propose that specifically targeting the exaggerated NLRP3 inflammasome response component in SSR asthma is a novel and viable therapeutic approach and suggest that this strategy is clinically preferable because it allows for IL-1β processing through other mechanisms for protection against infections [8, 30].
Biologics
There is ample recent evidence that next generation biologics may be steroid sparing or potentially be treatments for SSR disease. IL-5 is well known to be a potent signal for the development, maturation and chemotaxis of eosinophils [4, 5]. Numerous studies suggest that mepolizumab (anti-IL-5 monoclonal antibody) treatment reduces the requirement for oral steroids in severe eosinophilic asthma [96, 97]. Furthermore, there are known links between innate type 2 inducers and mediators IL-33, IL-25 and TSLP (thymic stromal lymphopoietin), and type 2 innate lymphoid and T-helper cells and steroid resistance [98–100]. Antibodies against these factors may prove to be somewhat effective in severe disease [4, 99, 101].
Conclusions
SSR asthma is a clinical problem of significant concern and more effective, and potentially broadly applicable, therapies are urgently required. As a consequence of the underlying mechanisms of pathogenesis, patients do not respond to mainstay steroid-based therapies. Steroid resistance in asthma has been linked to bacterial and viral respiratory infections, infection-induced exacerbations, and a high-fat diet/obesity. Understanding the mechanisms of pathogenesis will enable the identification of novel therapeutic approaches and the development of new and effective treatments. Collectively, clinical and experimental studies have identified macrolides and inhibitors of miR-21, PI3K and inflammasomes as potential new treatments for SSR asthma that may be broadly effective across multiple SSR asthma endotypes. Importantly, our data suggest that aberrant miR-21 and IL-1β responses drive steroid-resistant airway inflammation and AHR in SSR asthma through mechanisms that are independent of the canonical steroid response pathway and are therefore not amenable to steroid-mediated suppression. The logical extension of these studies would be to identify and characterise the roles, and assess the potential for therapeutic targeting, of other miR-21 targets and downstream mediators of IL-1β responses in SSR asthma. Biologics that target type 2 responses may also have utility in SSR asthma.
Footnotes
Provenance: Commissioned article, peer reviewed.
Conflict of interest: R. Wadhwa has nothing to disclose.
Conflict of interest: K. Dua has nothing to disclose.
Conflict of interest: I.M. Adcock has nothing to disclose.
Conflict of interest: J.C. Horvat has nothing to disclose.
Conflict of interest: R.Y. Kim has nothing to disclose.
Conflict of interest: P.M. Hansbro reports grants to develop inflammasome inhibitors for therapeutic use.
Support statement: R.Y. Kim is supported by a Fellowship from Lung Foundation Australia and Boehringer Ingelheim. P.M. Hansbro is supported by a Fellowship from the National Health and Medical Research Council of Australia (NHMRC #1079187).
- Received August 1, 2019.
- Accepted September 19, 2019.
- Copyright ©ERS 2019.
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










