





Abstract
Interstitial lung diseases (ILDs) are a set of heterogeneous lung diseases characterised by inflammation and, in some cases, fibrosis. These lung conditions lead to dyspnoea, cough, abnormalities in gas exchange, restrictive physiology (characterised by decreased lung volumes), hypoxaemia and, if progressive, respiratory failure. In some cases, ILDs can be caused by systemic diseases or environmental exposures. The ability to treat or cure these ILDs varies based on the subtype and in many cases lung transplantation remains the only curative therapy. There is a growing body of evidence that both common and rare genetic variants contribute to the development and clinical manifestation of many of the ILDs. Here, we review the current understanding of genetic risk and ILD.
Abstract
Common and rare genetic variants contribute to the development and clinical manifestation of many interstitial lung diseases http://bit.ly/31loHLh
Introduction
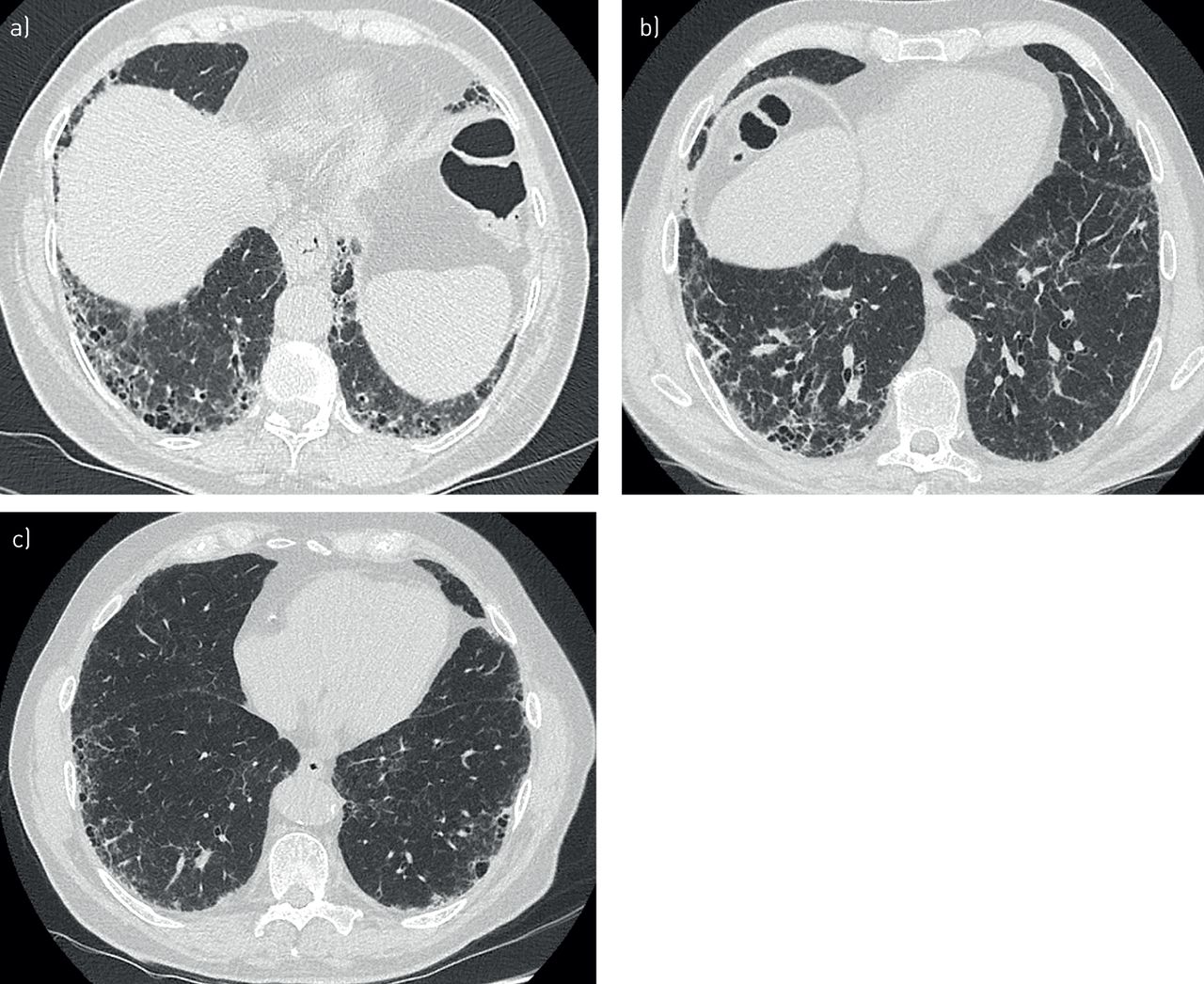
There is growing evidence that genetic factors contribute to the development of interstitial lung disease (ILD), notably in a context of familial aggregation (i.e. familial interstitial pneumonia (FIP)). Idiopathic pulmonary fibrosis (IPF) is the most common and most severe form of ILD, and has thus received the most attention in pulmonary research. High-resolution computed tomography (HRCT) of the chest in IPF patients shows interstitial fibrosis, described as the usual interstitial pneumonia (UIP) pattern (figure 1). Patients typically progress to hypoxaemia and respiratory failure, with most patients dying from the disease within 5 years of diagnosis [1, 2]. There are no curative therapies, but two drugs (nintedanib and pirfenidone) have been shown to slow disease progression [3, 4]. Lung transplantation is feasible for only a small percentage of IPF patients.

High-resolution computed tomography imaging of usual interstitial pneumonia: a) a patient with idiopathic pulmonary fibrosis (IPF) and carrier of rs35705950 within MUC5B, b) a patient with IPF and carrier of a TERC mutation and c) a patient with rheumatoid arthritis-interstitial lung disease and carrier of rs35705950.
The incidence of IPF is approximately 20 per 100 000 males and 13 per 100 000 females [5], but rising. Most individuals present aged 50–75 years. Many individuals can be diagnosed with UIP findings on HRCT of the chest, but when expert HRCT review is not definitive, patients are referred to lung biopsy for diagnosis. IPF patients have lung pathology consistent with a UIP pattern, characterised by interstitial fibrosis, honeycomb changes, fibroblastic foci and a paucity of inflammation [6, 7]. While the aetiology of IPF remains unknown, genetic discoveries in monogenic familial forms of the disease over the past three decades have led to significant insights into the role of inherited risk mutations in disease pathogenesis and in the understanding of the intimate mechanisms of lung fibrosis.
Methods for genetic testing are rapidly evolving and allow for several genes to be analysed together. From targeted next-generation sequencing (NGS) panels to whole-genome sequencing (WES), each approach has its own advantages and disadvantages that should be specifically considered. Briefly, for patients with suspected inherited pulmonary fibrosis, most genetic laboratories are using targeted NGS panels analysing three to 300 selected genes altogether, although a few laboratories perform WES. Sanger sequencing is still used for familial study or confirmation of the presence of a genetic variation.
Familial interstitial pneumonia
Although there is no consensus definition, in the research setting FIP is usually defined as a case of ILD in which the patient also has a family history of two or more relatives with ILD [8, 9]. Early studies suggested that familial forms of the disease accounted for 2–4% of IPF [10, 11], although later evidence suggests that this percentage may be higher [12, 13]. Adults with FIP are essentially indistinguishable from sporadic IPF patients in terms of clinical presentation, radiographic findings and histopathology, except that those with FIP tend to present at earlier age [14].
A study of 111 families with FIP, including 309 individuals with ILD and 360 unaffected relatives, revealed that male sex (55.7% versus 37.2%; p<0.0001), age (68.3 versus 53.1 years; p<0.0001) and having ever smoked cigarettes (67.3% versus 34.1%; p<0.0001) were risk factors for developing ILD. A UIP pattern was identified in 85% of patients; however, pathological heterogeneity was observed within individual families: 45% of these families having two or more pathological patterns identified within the affected individuals, with numerous families having evidence of UIP and non-specific interstitial pneumonia (NSIP) histopathology [14], an observation consistent with previous findings [15, 16], suggesting that distinct ILD categorisations may share similar pathogenesis pathways. The identification of cigarette smoking as a FIP risk factor also suggested that the interplay between genetic predisposition and environmental exposures is central to familial disease [14]. Many analyses of FIP families have suggested an autosomal dominant mode of inheritance with incomplete penetrance [11, 14, 17].
Common variants associated with ILD
Common variants associated with ILD are listed in table 1.
Common variants associated with idiopathic pulmonary fibrosis
MUC5B
In 2011, Seibold et al. [9] identified that a single nucleotide polymorphism rs35705950 is located in the promoter region of MUC5B, which encodes mucin 5B, a glycosylated macromolecular component of mucus, and is expressed in the normal bronchiolar epithelium. Using case–control analysis of non-Hispanic Whites, Seibold et al. [9] observed that subjects heterozygous and homozygous for the T risk allele had increased ORs for disease of 6.8 (95% CI 3.9–12.0) and 20.8 (95% CI 3.8–113.7) for FIP and 9.0 (95% CI 6.2–13.1) and 21.8 (95% CI 5.1–93.5) for IPF, respectively. This initial study found a similar rs35705950 minor allele frequency between FIP and sporadic IPF individuals (0.338 and 0.375, respectively), suggesting a similar genetic contribution of the MUC5B risk variant in sporadic and familial IPF [9]. Microscopy of diseased lung also reveals that MUC5B protein is found in the honeycomb cyst, a characteristic pathological finding of UIP, the pattern consistent with IPF [18].
Numerous groups have replicated the association between MUC5B rs35705950 and IPF, identifying this variant as the strongest and most well-replicated single genetic risk factor for IPF [9, 19–25]. The rs35705950 T risk allele is common and was detected in 10% of a non-Hispanic White control population [26]. The presence of the variant alone is insufficient to cause disease and approximately half of subjects with IPF do not carry this variant. The rs35705950 variant is neither necessary nor sufficient to cause disease, suggesting the involvement of other genetic or environmental factors to cause disease development; this remains an area of active research [27].
The MUC5B promoter polymorphism also appears to be specific to the risk of UIP and then most frequently associated with IPF, but eventually also associated with a UIP pattern in the context of hypersensitivity pneumonitis or rheumatoid arthritis (RA)-ILD [28, 29]. Indeed, in a study including 620 RA-ILD patients, 614 RA without ILD patients and 5448 unaffected controls, the MUC5B risk polymorphism was associated with the risk of ILD in RA patients when compared with unaffected controls or with RA without ILD patients. Interestingly, the increased risk of ILD was restricted to patients with a UIP pattern (41% of the whole RA-ILD group): UIP RA-ILD versus RA-no ILD (OR 6.1, 95% CI 2.9–13.1; p=2.5×10−6) and non-UIP RA-ILD versus RA-no ILD (OR 1.3, 95% CI 0.6–2.8; p=0.46) [29].
Moreover, in two cohorts of 145 and 72 Caucasian chronic hypersensitivity pneumonitis patients, the MUC5B risk polymorphism prevalence was 24.4% and 32.3%, respectively, versus 10.7% in the general population [28]. Among the 189 patients with chronic hypersensitivity pneumonitis and CT scan available, the MUC5B risk polymorphism was associated with the presence of traction bronchiectasis, suggestive of probable UIP (p<0.001), but not with a pattern consistent with definite or possible UIP or presence of radiographic honeycombing [28]. rs35705950 has not been associated with ILD in systemic sclerosis [20, 22], sarcoidosis [20] or inflammatory myositis, or with chronic obstructive pulmonary disease or asthma [30]. A subsequent genome-wide association study (GWAS) that examined numerous genetic loci as well as rs35705950 in a fibrotic idiopathic interstitial pneumonia (IIP) cohort that largely contained IPF subjects, but also contained other forms of fibrotic IIP, confirmed the association between the MUC5B genotype and the fibrotic IIP phenotype [31].
rs35705950 has also been associated with a risk of IPF in Hispanic White and Asian populations, although the overall rs35705950 frequency is low in Asian populations [24, 25, 32]. IPF is thought to be rare in African populations [33].
Other common genetic variants and IPF
Although the common MUC5B promoter polymorphism is the most widely and well-studied common genetic variant associated with IPF and FIP, other common variants have been discovered through GWAS as high-throughput variant screening methods have developed.
In 2008, researchers from Japan identified an association of a common TERT (telomerase reverse transcriptase) variant with susceptibility to IPF [34]. In 2013, a large GWAS confirmed several known disease-associated loci (chromosome 5p15 which contains TERT; 11p15 which contains MUC5B; 3q25 near TERC (telomerase RNA component)) and identified seven new loci, including FAM13A (family with sequence similarity 13 member A; 4q22), DSP (desmoplakin; 6p24), OBFC1 (oligonucleotide-binding fold containing 1; 10q24), ATP11A (ATPase phospholipid transporting 11A; 13q4), DPP9 (dipeptidyl peptidase 9; 19p13), and risk loci on chromosomes 7q22 and 15q14–15 [31]. The implicated genes span a wide variety of biological functions, but could be categorised into the following: host defence (MUC5B and ATP11A), cell–cell adhesion (DSP and DPP9) and DNA repair (TERT, TERC and OBFC1) [31, 35–37]. It has been estimated that these loci, excluding the MUC5B variant, may account for up to a third of disease risk, emphasising the importance of genetic predisposition in fibrotic ILD [31, 37].
Another IPF GWAS performed by an independent group replicated the MUC5B rs35705950 association, but also suggested the contribution of additional risk alleles located in TOLLIP (Toll-interacting protein) and SPPL2C (signal peptidase-like 2C). Importantly, this study not only identified risk variants, but also drew connections between specific variants (rs5743890) in TOLLIP and differential mortality from disease [21]. However, a recent study suggested that rs5743890 in TOLLIP was not associated with increased risk of IPF when adjusted for the presence of other genetic risk factors such as MUC5B [38].
Another GWAS identified a new locus associated with IPF near AKAP13 (A-kinase anchoring protein 13; rs62025270, OR 1.27, 95% CI 1.18–1.37; p=1.32×10−9). The allele associated with increased susceptibility to IPF was also associated with increased expression of AKAP13 mRNA in control lung tissue [39]. Interestingly, as was observed in the initial MUC5B promoter polymorphism study [9], the odds ratios for loci identified by the 2013 GWAS by Fingerlin et al. [31] did not differ between FIP and sporadic IPF cases, reinforcing that both diseases share a common genetic background.
Disease severity
Retrospective analyses of large clinical trials data reveals that IPF patients with the minor allele (T) at rs35705950 in MUC5B had improved survival when compared with wild-type (GG) subjects of the same cohort [40], suggesting that the MUC5B promoter variant identifies a subset of patients with IPF who have a distinct phenotype/prognosis. Similarly, genotype at the variant in TOLLIP first associated with IPF by Noth et al. [21] (rs5743890) is also associated with differential survival and may be associated with a differential response to N-acetylcysteine [41].
A post hoc analysis of the CAPACITY and ASCEND trials showed that patients with the MUC5B risk allele were older (68.1 versus 65.5 years) and had a slower disease progression than patients without the risk allele. Pirfenidone was, however, still associated with a decreased decline of forced vital capacity (FVC) [42]. Nintedanib has not been examined in terms of efficacy by genotype.
Rare variants associated with ILD
Numerous Mendelian disorders can be associated with ILD. Rare variants associated with ILD are listed in table 2. Here, we will focus on the most frequent causes: surfactant-associated protein gene mutations and telomere-related genes.
Rare variants associated with interstitial lung disease
Surfactant proteins
SFTPC
Surfactant protein C (SPC) is one of four surfactant proteins expressed in the alveoli and functions to alter surface tension to prevent alveolar collapse. This protein is expressed throughout the lung epithelium during lung development, but in the mature lung it is localised to type II alveolar epithelial cells [43]. Early studies of genetic risk in the development of IPF used FIP subjects. The first disease-associated genetic variants were identified in surfactant protein genes among FIP patients [15, 44–46]. These studies identified heterozygous mutations in SFTPC coding for SPC [15, 44], which segregated with diseased subjects and was not found to be present in controls.
Both paediatric and adult ILD have been linked to SFTPC mutations [15]. Although SFTPC mutations were first linked to paediatric cases of ILD, the contribution of SFTPC mutations in adult FIP has also been established. In 2002, Thomas et al. [16] described a family in which 11 adults had ILD, six with biopsy-confirmed UIP/IPF and five with clinical diagnoses of IPF, as well as three paediatric cases of NSIP. In vitro studies also revealed that the L188Q SFTPC mutation results in a pro-SPC molecule that cannot be folded properly, prompting endoplasmic reticulum stress and caspase pathway activation [47, 48]. Subsequently, additional mutations in SFTPC have been found in other FIP cohorts, up to 25% of FIP cases in a Dutch cohort, although this is a lot higher than what has been observed elsewhere [49, 50].
SFTPC mutations are rarely found in sporadic IPF cases. Interestingly, de novo mutations are frequent in children and may explain up to 50% of cases [51].
Other surfactant-related genes
Heterozygous mutations in SFTPA2 (surfactant protein A2) or SFTPA1 (surfactant protein A1) have been identified in subjects with FIP and/or lung adenocarcinoma [46, 52].
ATP-binding cassette transporter A3 (ABCA3) is expressed in type II alveolar epithelial cell lamellar bodies and is important in surfactant processing. Although homozygous ABCA3 mutations are usually associated with respiratory failure in newborns [53], one teenage ILD patient with a UIP pattern and one 41-year-old patient with combined pulmonary fibrosis and emphysema (CPFE) carrying mutations of ABCA3 have been reported [54, 55]. Other studies have suggested that in infant ILD, those with heterozygous SFTPC mutations and concomitant heterozygous mutations in ABCA3 may be more likely to develop clinical ILD [56]. Therefore, ABCA3 recessive mutations may modify the effects of SFTPC dominant mutations [57].
NKX2-1 (NK2 homeobox 1) encodes a transcription factor closely related to surfactant protein transcription [58]. Heterozygous mutations are classically associated with the triad of ILD, hypothyroidism and neurological anomalies (hypotonia, delayed development and chorea) [59]. These mutations may be associated with ILD without hypothyroidism and neurological anomalies in up to a third of cases, including adult cases in which the most common HRCT pattern is atypical for UIP [59].
Biallelic ABCA3 mutations and heterozygous NKX2-1, SFTPA1 SFTPA2 and SFTPC mutations in adults may share similar clinical and radiological presentation. The most frequent radiological pattern associates predominant diffuse ground-glass opacities, septal thickening and cysts of variable size with a preferential distribution in the upper lobes and in subpleural areas (figure 2). Differentiating emphysema from cysts is sometimes difficult and SFTPC mutation must be evoked in a young patient presenting CPFE [60]. However, at a later stage of disease, honeycombing can predominate.

{kind=link}
{kind=link}
High-resolution computed tomography of a) non-usual interstitial pneumonia (UIP) pattern (indeterminate) with ground-glass opacities and reticulation associated with a SFTPA1 mutation, and b, c) non-UIP pattern (indeterminate) with ground-glass opacities and cysts from two patients, both carriers of a compound heterozygous ABCA3 mutation. d) Pattern suggestive of pleuroparenchymal fibroelastosis associated with a TERT mutation.
Histologically, the most frequently related pattern in adults is UIP, although NSIP, organising pneumonia or desquamative interstitial pneumonia have also been reported. Moderate inflammation and centrolobular fibrosis can be observed [50].
In children, successful treatments reported in case reports or short series include methylprednisolone, hydroxychloroquine or azithromycin [61–63]. No treatment appears to reduce disease in a patient with predominant honeycombing lesions. The effect of antifibrotic drugs, such as pirfenidone or nintedanib, is to date unknown. The disease does not appear to recur after pulmonary transplantation [61].
Telomere-related genes
Telomeres are regions of noncoding repetitive nucleotide repeats (TTAGGG) at the ends of chromosomes that protect them from deterioration during mitosis or fusion with neighbouring chromosomes. The telomerase complex is the group of proteins and RNA that catalyses the addition of these nucleotide repeats to the ends of chromosomes. There are numerous components to the telomerase complex, including TERT and TERC, which are essential for normal operation and telomere integrity. Shortening of telomeres has been associated with numerous disease manifestations, as have mutations in telomere-related genes [64], including ILD. Indeed, numerous studies of FIP cases and their kindred have identified mutations in various telomere-related genes (TERT, TERC, RTEL1 (regulator of telomere elongation helicase 1), PARN (poly(A)-specific RNase), NAF1 (nuclear assembly factor 1 ribonucleoprotein), DKC1 (dyskerin pseudouridine synthase 1) and TINF2 (TERF1 interacting nuclear factor 2)). For instance, TERT and TERC mutations have been identified in up to a sixth of pulmonary fibrosis families [65, 66].
Dyskeratosis congenita (DKC) is a diagnosis made based on a triad of abnormal skin pigmentation, nail dystrophy and oral leukoplakia, but can affect numerous organ systems [67], including the bone marrow. Pulmonary fibrosis is found in 20% of cases and respiratory failure is the most common proximal cause of death in these patients. In X-linked DKC, mutations in DKC1 are causative [68, 69], but some autosomal dominant forms of DKC are linked to mutations in TERT and TERC [70–72]. In 2005, Armanios et al. [72] reported a TERT mutation in a family affected by DKC in which pulmonary fibrosis was the dominant clinical finding.
Subsequently, Armanios et al. [72] and Tsakiri et al. [66] identified heterozygous TERT and TERC mutations. In vitro examination of the mutations demonstrated decreased telomerase activity and that peripheral blood leukocyte telomere lengths were shorter in mutation carriers when compared with age-matched non-carriers. These studies suggested that telomere-related gene mutations cause disease in ∼15% of FIP.
Armanios et al. [72], Tsakiri et al. [66] and others also examined telomere length itself and its relationship to pulmonary fibrosis, independent of mutations in TERT and TERC [73, 74]. Cronkhite et al. [73] analysed a cohort of pulmonary fibrosis patients without TERT and TERC, including probands from 59 families with FIP and 73 subjects with sporadic IPF. They found that 24% of FIP subjects and 23% of sporadic IPF subjects had evidence of telomere shortening, with peripheral blood leukocyte telomere lengths below the 10th percentile compared with age-matched controls. Alder et al. [74] analysed 100 cases of sporadic IPF, and found one subject with a TERC mutation and no mutations in TERT. 62 of these subjects had their telomere lengths measured in peripheral blood lymphocytes and 97% showed telomere lengths shorter than the median in healthy controls; furthermore, 10% had telomere lengths shorter than the first percentile of healthy controls.
Alder et al. [74] found cryptogenic cirrhosis in a few of the IPF subjects, which prior to their publication had only been described in the setting of DKC. These additional findings suggested that, at least in a small subset of patients, “telomeropathy”, or a syndrome in which multiple organs are affected by telomere shortening, may be present. A subsequent study that examined this link further sequenced numerous subjects with both aplastic anaemia and pulmonary fibrosis, and found that the concurrence of these two disorders (both separately associated with telomere dysfunction) was highly predictive for the presence of germline telomerase mutation [75, 76], a finding that could affect the clinical evaluation and decision making for those contemplating bone marrow or lung transplantation.
More recent studies have utilised WES techniques to discover rare variants in other telomerase pathway genes. Specifically, this technique has been utilised to pinpoint rare variants in the RTEL1 and PARN genes found to be associated with FIP [77–79]. As in the case of other telomerase pathway genes, affected subjects with the identified genetic variants in these genes had evidence of shortened peripheral blood leukocyte telomeres [77, 78], although the mechanism through which PARN mutations affect telomere length remains poorly understood [80]. Exome sequencing has also identified rare TINF2 and NAF1 mutations in FIP [81, 82]. Additionally, a novel DKC1 mutation was also recently described in association with FIP [83].
Mechanistically, although the specific link between telomere-related gene mutations and pulmonary fibrosis remains an area of active research, in vivo studies utilising mouse models for loss of function of telomere-related genes suggest that when these genes dysfunction, the lung epithelia's response to injury is impaired [84].
Heterozygous mutations have been detected in familial forms of pulmonary fibrosis involving TERT (∼15%), RTEL1 (5–10%), PARN (∼5%) and TERC (∼3%). Mutations in DKC1, NAF1 and TINF2 are much rarer [76–79, 81, 83, 85, 86]. Telomere-related gene mutations may be found in 1–9% of sporadic IPF cases [42, 87]. None of these genes is the site of a frequent mutation and new genetic variants are continually being identified. The penetrance (risk of pulmonary fibrosis developing in a telomere-related gene mutation carrier) depends on several factors, including environmental exposure [76].
Telomeres shorten from generation to generation in patients with TERT, TERC or RTEL1 mutations because of transmission of the short telomeres independent of transmission of the mutation [86]. As telomere shortening varies depending on the involved gene and its impact on telomere length, genetic anticipation may be more pronounced for carriers of TERC than PARN mutations [86].
The disease phenotype of patients carrying telomerase pathway mutations is varied. In individuals with known telomerase pathway mutations, the prevalence of ILD increases with age, as illustrated by a study of TERT mutation carriers in which none of the subjects <40 years of age had evidence of ILD, yet its prevalence in those >60 years of age was 60% [88]. An observational study of 115 pulmonary fibrosis patients with telomerase pathway mutations (TERC, TERT, RTEL1 and PARN) was conducted and found that TERC mutation carriers were diagnosed at an earlier age (mean 51 years) relative to the other study subjects (58 years for TERT, 60 years for RTEL1 and 65 years for PARN) [86].
Pulmonary fibrosis and extrapulmonary manifestations associated with telomerase complex mutations are variably called “telomere disease”, “telomeropathy” or “short telomere syndrome”, with lack of a consensual definition. In patients with TERT mutation and pulmonary fibrosis, the classical triad of DKC is usually absent. However, 15–40% of mutation carriers present premature hair greying (before 30 years of age) [76, 89]. In patients with telomere-related gene mutations and pulmonary fibrosis, anaemia is present in 17–27%, macrocytosis in 24–41% and thrombocytopenia in 8–54% [76, 86, 88]. DKC1, TINF2 and TERC mutation carriers seem more prone to the development of haematological involvement than TERT or RTEL1 mutation carriers (our own observation and [86]). Patients can also present liver cirrhosis: cryptogenic, viral or alcoholic. Elevated liver enzyme levels or liver involvement was reported in 5–27% of patients with ILD and telomere-related gene mutations [76, 86]. Gorgy et al. [90] highlighted the high frequency of hepatopulmonary syndrome associated with telomere-related gene mutations in a retrospective series of nine patients without ILD. Among the six patients with available liver biopsies, the most common abnormality was nodular regenerative hyperplasia (in four patients) [90].
A typical UIP pattern on chest CT was initially reported in up to 74% of cases, but was recently found in only 46–55% of cases (figure 1) [76, 86, 88]. Unusual features found in 13–20% of cases included upper lung predominance of fibrosis, centrolobular fibrosis or a pleuro-parenchymal fibroelastosis pattern (figure 2) [76, 86, 88, 91]. Taken together, 14–40% of cases show a CPFE pattern [92].
The decline of pulmonary function (FVC) of patients with ILD associated with telomerase complex mutations seems unexpectedly high when compared with placebo arms of IPF clinical trials (130–210 mL per year) [93]. Newton et al. [86] reported a 300 mL per year decline of FVC whatever the gene involved (TERC, TERT, RTEL1 or PARN) and the ILD entity (IPF or not).
The safety and effectiveness of pirfenidone has been reported in patients with telomere-related gene variants. One European retrospective study was not able to show an effect of pirfenidone on lung function decline, with a decline of FVC of 161.8±31.2 mL per year before and 235.0±49.7 mL per year after pirfenidone initiation [94]. However, a post hoc analysis of two phase 3 clinical trials (CAPACITY and ASCEND) identified 102 patients with IPF as carriers of rare telomere-related gene variants. Although carriers of a rare variant within TERT, PARN, TERC or RTEL1 had a more rapid decline in predicted FVC than patients without a rare variant (1.66% versus 0.83% per month), pirfenidone still reduced the decline of FVC in this subgroup of severe patients [42]. No data are available for nintedanib.
Danazol, a synthetic sex hormone with androgenic properties, showed promise for pulmonary fibrosis associated with telomere disease, with stabilisation of diffusing capacity of the lung for carbon monoxide, FVC and CT scan findings during a 2-year treatment period [95].
Given the young age of most patients, lung transplantation is often discussed. At least five retrospective series reported the outcome of lung transplantation in 61 telomere-related gene mutation carriers [96–100]. Most patients required adjustment of immunosuppression because of haematological toxicity. Thrombocytopenia and a need for platelet transfusion were frequent, and myelodysplastic syndrome and/or bone marrow failure occurred in some patients. Acute kidney failure requiring dialysis support seemed unexpectedly frequent (up to 50%) [96, 97]. Interestingly, short telomeres and mutations of telomere-related genes have been associated with increased prevalence of cytomegalovirus (CMV) infection after lung transplantation [100]. Very recently, in a cohort of 262 patients who received lung transplantation, patients with TERT, RTEL1 or PARN mutations (n=31 (11.8%)) were reported to have a reduced post-transplantation survival (hazard ratio 1.82, 95% CI 1.07–3.08; p=0.03) and higher risk of chronic lung allograft dysfunction (hazard ratio 2.88, 95% CI 1.42–5.87; p=0.004) [99]. However, this retrospective study did not report higher risk of haematological complication or renal insufficiency in telomere-related gene mutation carriers [99].
In an independent cohort, patients with telomere length below the 10th percentile before transplant were reported to have a worse survival and also a shorter time to onset of chronic lung allograft dysfunction [101]. Comparison of the less than 10th percentile telomere length group with the greater than 10th percentile telomere length group showed a higher rate of primary graft dysfunction, but there were no differences in the incidence of acute rejection, cytopenias, infection or renal dysfunction [101].
However, in some patients, telomere-related gene mutations have also been associated with a risk of immunodeficiency with a spontaneous risk of opportunistic infection such as Pneumocystis jirovecii or after lung transplantation as assessed by an increased risk of CMV infection [100, 102].
Combined rare and common variants
The studies described in detail in the previous sections have generally taken the approach of examining either common variants or rare variants and their relationship to IIP or IPF risk [76, 103]. Very recently, a combined analysis of rare and common variants of 1510 patients with IPF showed 1046 patients (69.2%) were carriers of the rs35705950 (MUC5B) risk allele, but only 30 (3%) of them were also carriers of a rare variant within TERT, whereas 34 (7%) of the non-carriers of rs35705950 were also carriers of a rare variant within TERT [42]. Furthermore, in a recently reported study of 3624 IPF patients and 4442 controls, deep targeted resequencing of candidate genes showed that TERT and RTEL1 were independently associated with the risk of IPF [38].
Conclusions
Although these initial studies suggest that genetic variants could be useful in assisting with making a prognosis, the relationships between genotype at different variants and survival are still being investigated and need to be validated in prospective studies. Future therapeutic trials will need to take into account phenotypic and genotypic variation to allow for a deeper understanding of how these characteristics can and should be integrated into shared decision making. At present, given the limited data definitively linking genetic variants with concrete clinical outcomes or therapeutic responses, sequencing and genotyping patients are not part of routine IPF or fibrotic IIP care. Our point actually is to consider genetic analysis (including telomere-related gene and MUC5B sequencing) and telomere length for familial pulmonary fibrosis, short telomere syndrome, and sequencing surfactant genes for cryptogenic pulmonary fibrosis below the age of 50 years. Evidence of a pathogenic mutation should at least lead to genetic counselling while awaiting targeted therapy [104].
Footnotes
Provenance: Publication of this peer-reviewed article was sponsored by Boehringer Ingelheim, Germany (principal sponsor European Respiratory Review issue 153).
Conflict of interest: R. Borie reports personal fees and non-financial support from Roche, and Boehringer Ingelheim, outside the submitted work.
Conflict of interest: P. Le Guen has nothing to disclose.
Conflict of interest: M. Ghanem has nothing to disclose.
Conflict of interest: C. Taillé has received personal fees and other funding from AstraZeneca and Roche, personal fees from Teva and Genzyme, grants, personal fees and other from GlaxoSmithKline, Novartis and Sanofi, and other funding from Boehringer Ingelheim.
Conflict of interest: C. Dupin reports personal fees, non-financial support and other from AstraZeneca, Boehringer, GlaxoSmithKline and Novartis, personal fees and other from Chiesi, personal fees from Sanofi, and non-financial support and other from Roche, outside the submitted work.
Conflict of interest: P. Dieudé has nothing to disclose.
Conflict of interest: C. Kannengiesser has nothing to disclose.
Conflict of interest: B. Crestani reports personal fees from AstraZeneca, grants, personal fees and non-financial support from Boehringer Ingelheim and Roche, personal fees and non-financial support from Sanofi, and grants from Novartis, outside the submitted work.
- Received May 24, 2019.
- Accepted August 1, 2019.
- Copyright ©ERS 2019.
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










