





Abstract
Idiopathic pulmonary fibrosis is a fatal age-related lung disease characterised by progressive and irreversible scarring of the lung. Although the details are not fully understood, there has been tremendous progress in understanding the pathogenesis of idiopathic pulmonary fibrosis, which has led to the identification of many new potential therapeutic targets. In this review we discuss several of these advances with a focus on genetic susceptibility and cellular senescence primarily affecting epithelial cells, activation of profibrotic pathways, disease-enhancing fibrogenic cell types and the role of the remodelled extracellular matrix.
Abstract
This review provides a summary of the most important findings in basic science investigations in pulmonary fibrosis and how they affect drug development and future patient management. http://bit.ly/2RjGMFZ
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic fibrosing interstitial lung disease with a median survival time of 3–5 years after diagnosis [1, 2]. It is an age-related disease, with the vast majority of individuals being diagnosed at >60 years of age [1]. IPF is associated with exertional dyspnoea, chronic cough, declining lung function and impairment in quality of life. Many patients with IPF experience acute exacerbations and acute episodes of respiratory worsening, associated with up to 50% mortality rate [3, 4].
Formerly regarded as a result of chronic inflammation, clinical trials with a combination of anti-inflammatory drugs (prednisone, azathioprine and N-acetyl-l-cysteine) failed to improve outcomes [5]. In recent decades, though not fully understood in detail, there has been tremendous progress in understanding the pathogenesis of IPF. The current hypothesis is that subclinical alveolar epithelial injury imposed on ageing epithelial cells in genetically susceptible individuals leads to aberrant wound healing, secretion of high levels of growth factors, cytokines, chemokines, accumulation of fibroblasts and differentiation into myofibroblasts, and deposition of the extracellular matrix (ECM). To date, two drugs for IPF are available, pirfenidone and nintedanib, both slowing disease progression [6]. Other investigational therapies currently target many of the mediators and signalling pathways involved in the pathogenesis of pulmonary fibrosis (table 1) [7].
Key targets in fibrogenesis and their corresponding clinically tested drugs
In this review, we highlight some of the current knowledge of the pathogenesis of IPF from basic science and describe how these may have an impact on potential future therapies for this devastating lung disease.
Genetic susceptibility
For decades, case series of pulmonary fibrosis occurring in families suggested a genetic predisposition to this disease. These heritable pulmonary fibrosis forms are called familial interstitial pneumonia (FIP) and have been investigated in detail to try and understand the pathogenesis of IPF. A breakthrough occurred in 2001, when Nogee et al. [8] identified a heterozygous mutation in the SFTPC gene, encoding surfactant protein (SP)-C. Subsequently, additional mutations in SFTPC were reported [9–11]. Furthermore, mutations in SFTPA2, encoding SP-A2 in FIP and sporadic IPF, and mutations in SFTPA1, encoding SP-A1 in FIP, were identified [12–14]. Expression of mutant SFTPC proteins in human alveolar epithelial cells (AECs) led to the aggregation of mutated SP-C protein in the endoplasmic reticulum and increased endoplasmic reticulum stress [15]. Transgenic mice expressing mutant L188Q SP-C found in FIP, exclusively in type 2 AECs, showed endoplasmic reticulum stress and exaggerated lung fibrosis after bleomycin administration [16]. Transgenic mice expressing mutant I73T SFTPC in type 2 AECs developed spontaneous lung fibrosis [17]. These findings indicate that endoplasmic reticulum stress in type 2 AECs, increased by the mutant SP-C protein, contributes to fibrogenesis. Similarly, SP-A2 mutant proteins (G231V and F198S) remaining within the endoplasmic reticulum enhance endoplasmic reticulum stress [12, 18]. SP mutations reportedly account for no more than 5% of sporadic IPF but considering that both SP-A and SP-C are produced exclusively by type 2 AECs, these studies still provide strong evidence that recurrent epithelial cell injury is a major factor in IPF pathogenesis.
In 2011, a large genome-wide linkage study of patients with IPF identified a common single nucleotide polymorphism (SNP) (rs35705950) in the promoter region of MUC5B associated with a 20-fold increased risk of IPF in subjects who were homozygous for the SNP (seven-fold in heterozygous subjects) [19]. These findings have been replicated in several studies since the first report [20–23]. While at least one copy of the variant seems to be present in 34–38% of patients with IPF, it also is present in 9% of healthy controls. Interestingly, the rs35705950 variant is associated with some forms of fibrotic interstitial lung diseases (rheumatoid arthritis-related interstitial lung disease and chronic hypersensitivity pneumonitis) [24, 25] but not associated with a variety of other fibrotic interstitial lung diseases (scleroderma-related interstitial lung disease, asbestosis, sarcoidosis) [22, 23, 26], suggesting a specific role for the variant in the pathogenesis of pulmonary fibrosis. The frequencies of the T-allele at rs35705950 is different among ancestries (European: 11%, South Asian: 8%, East Asian: 1%, African: <1%) [27]; the risk associated with the rs35705950 variant among Asians [28, 29] and Mexicans [30] is similar to the risk among people with European ancestry. This is important when the variant is considered as a tool for early diagnosis [31] or as a prognostic marker [32].
MUC5B encodes mucin 5B, a glycoprotein required for airway clearance and innate immune responses to bacteria [33]. The risk allele at rs35705950 is a gain-of-function variant associated with overexpression of mucin 5B in small airway epithelial cells [34] and honeycomb cysts in IPF lungs [35]. In 2018, Hancock et al. [36] generated two lines of transgenic mice that overexpressed mucin 5B in airways/type 2 AECs and showed that overexpressing mucin 5B caused impaired mucociliary clearance and augmented bleomycin-induced lung fibrosis but did not trigger spontaneous fibrosis. Despite these interesting findings, it is still unclear how one can explain the fact that individuals with this risk allele within a group of patients with IPF have a better prognosis than individuals who do not have it [32].
Other recently reported genetic variants in DSP, AKAP13, CTNNA and DPP9 that are responsible for cell adhesion, integrity and mechanotransduction also increase the risk of IPF [37, 38]. From a biological perspective, all these genetic mutations and variants probably predispose individuals to AEC dysfunction following epithelial injury at the initiation of fibrosis.
Cellular senescence
Ageing is an important risk factor for IPF and senescence of AECs seems to be a central phenotype that promotes pulmonary fibrosis [39]. Shortened telomeres, endoplasmic reticulum stress and mitochondrial dysfunction lead to AEC senescence. Specific signalling pathways in cellular senescence can be targeted as novel therapeutic interventions.
Telomeres in IPF
Pulmonary fibrosis has been found in some patients with dyskeratosis congenita (DKC), a disorder that is inherited and characterised by skin hyperpigmentation, nail dystrophy and aplastic anaemia. DKC is most commonly caused by mutations in DKC1, a component of the telomerase complex, but mutations in genes encoding other members of the telomerase complex (TERT and TERC) have also been reported [40]. In a cohort with FIP, 8% of patients had heterozygous mutations in TERT or TERC, and mutation carriers had short telomeres [41], suggesting that mutations in TERT and TERC can cause pulmonary fibrosis. Furthermore, since manifestations of DKC reflect aberrant epithelial cell function, the finding of telomere mutation in FIP underscores the importance of AECs in the pathogenesis. Additional evidence, suggests that telomere dysfunction in IPF derives from the fact that abnormal telomere shortening is not exclusive to patients with telomerase mutation-associated pulmonary fibrosis [42–45]. In the past few years, several potential therapeutic strategies to potentiate telomerase activity have been proposed, especially the usage of oestrogen and androgens. Androgens can restore telomerase activity in circulating leukocytes and haematopoietic stem cells from subjects with reduced telomerase function associated with TERT mutations [46, 47]. Based on this evidence, an early-phase clinical trial with the synthetic androgen, danazol, was performed in patients with short telomeres, most of whom had pulmonary fibrosis, and showed that telomeres can be lengthened by this intervention [48].
Mitochondrial dysfunction
Mitochondrial dysfunction contributes to the pathogenesis of several age-related diseases, including IPF [49, 50]. In IPF lungs, AECs exhibit large numbers of damaged mitochondria [51], and increased levels of free mitochondrial DNA were found in the plasma and bronchoalveolar lavage of patients with IPF [52]. Special attention has been paid to mitophagy in regulating cell fate for both AECs and fibroblasts. Mitophagy describes the selective lysosomal degradation of damaged mitochondria and is mainly governed by a signalling molecule called PINK1 [53]. Decreased expression of PINK1 in type 2 AECs was found in IPF lungs and correlated with the accumulation of dysmorphic mitochondria and increased AEC apoptosis [51]. In 2018, Yu et al. [54] reported that the activity and expression of iodothyronine deiodinase 2 (DIO2), an enzyme that activates thyroid hormone, were higher in IPF lungs. Dio2-deficient mice exhibited enhanced bleomycin-induced lung fibrosis and delivery of aerosolised thyroid hormone resolved fibrosis and improved survival. The treatment with thyroid hormone promoted the expression of PINK1, resulting in the restoration of normal mitochondrial function and rescue from mitochondria-regulated apoptosis [54]. These exciting findings suggest that promotion of mitophagy may reduce mitochondrial damage and could become a novel therapeutic strategy.
Profibrotic mediators
Dysfunctional AECs produce numerous mediators that promote migration of cells (such as fibrocytes, monocytes and fibroblasts) and induce their differentiation into fibrogenic cell types such as myofibroblasts. These fibrogenic cells are responsible for the accumulation of excessive amounts of ECM which eventually destroys the lung architecture. Transforming growth factor (TGF)-β, connective tissue growth factor (CTGF), platelet-derived growth factor (PDGF), endothelin-1, vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF) and CXC chemokine ligand 12 (CXCL12) are all secreted by type 2 AECs and promote profibrotic responses.
Among these factors, TGF-β is the most potent profibrotic mediator. TGF-β promotes AEC apoptosis, epithelial-mesenchymal transition, production of other profibrotic mediators, recruitment of circulating fibrocytes and fibroblast transformation into myofibroblasts [55]. CTGF is a matricellular protein that mediates tissue remodelling and fibrosis, acting to promote fibroblast migration, the formation and activation of myofibroblasts and ECM deposition [56, 57]. Recently a clinical trial with FG-3019, a monoclonal anti-CTGF antibody, showed a slower decline in lung function for patients with IPF [58]. PDGF is a growth factor known to be a strong stimulus of proliferation, migration and survival of fibroblasts produced by AECs and macrophages [59, 60]. The FGF family has 22 structurally related members, interacting with heparin sulfate glycosaminoglycans, and binding to extracellular FGF receptors (FGFRs) [61]. FGFs and FGFRs also have very important roles in cell proliferation, differentiation, migration and survival [62]. Particularly, FGF-2 is a mitogen for fibroblasts and induces collagen synthesis [63]. In IPF lungs, FGF-2 is produced by alveolar macrophages, fibroblasts, endothelial cells and mast cells and increased FGF-2 levels are present in IPF lungs [64]. The VEGF family consists of five secreted members, with VEGF-A and -B playing an important role in the regulation of blood vessel growth, while VEGF-C and -D mainly affect lymphangiogenesis [65]. VEGFs bind to the three different VEGF receptors (VEGFRs). VEGF-A has been shown to stimulate PDGF receptor (PDGFRs), thereby regulating mesenchymal cell migration and proliferation [66]. Inhibition of VEGFR may reduce experimental fibrosis [67]. All these findings highlight a complex interaction between these growth factors, which is dysregulated in fibrotic lungs. Targeting just one of them may not result in the expected therapeutic effect, which may explain why several clinical trials with single targets did not report more positive results [7].
Nintedanib is a potent inhibitor of several receptor tyrosine kinases for PDGFR, FGFR and VEGFR, and has shown efficacy in reducing the decrease of forced lung capacity [68]. Pirfenidone also targets several growth factors involved in fibrogenesis and has also shown efficacy on slowing the progression of IPF [69, 70]. Thus, targeting profibrotic mediators, ideally several and not just one, can have therapeutic benefits for IPF.
Disease modifying cells in IPF
Dysfunctional AECs can induce the migration and accumulation of profibrotic cells. The following section highlights recent findings in such disease-enhancing cells in IPF.
Fibroblasts
Fibroblasts are tissue mesenchymal cells that are key in establishing and maintaining a normal and structured ECM. During wound healing, epithelial cell activation and epithelial-mesenchymal signalling induce the migration and activation of fibroblasts, differentiation to myofibroblasts, deposition and remodelling of ECM. Too much or aberrant activity of these processes can turn wound healing into scarring and fibrosis. Lysophosphatidic acid has been identified as a mediator of fibroblast chemoattractant activity [71], and administration of GLPG1690, an inhibitor of autotaxin, the enzyme principally responsible for extracellular lysophosphatidic acid production, showed a slower decline in lung function for patients with IPF [72] and is currently undergoing phase III clinical trials (ClinicalTrials.gov identifier: NCT03711162 and NCT03733444).
Several mediators, including TGF-β and PDGF, can drive the differentiation of fibroblasts to myofibroblasts. Myofibroblasts secrete large amounts of ECM molecules, including collagen [73], and this excess production together with reduced removal of ECM leads to pathologic lung remodelling and fibrosis. Furthermore, IPF fibroblasts seem to obtain a specific phenotype with increased capacity for invasion [74, 75] and resistance to apoptosis [76, 77], making them more destructive and detrimental in the lungs. In 2019, Wohlfahrt et al. [78] reported that the transcriptional factor PU.1 was an essential regulator of profibrotic gene expression in fibroblasts. In theory, targeting such apoptosis-resistant fibroblasts could be a promising strategy for novel therapies.
Macrophages
Macrophages are a pool of tissue-resident and circulating cells that can transform from one phenotype to another [79, 80]. Their classical phenotypes are ‘M1’ or ‘M2’ macrophages [81]. The polarisation of macrophages is a very dynamic process in which macrophages develop various functional phenotypes in response to stimulation and signals from the microenvironment they live in [82]. Macrophages are the most abundant immune cells in the lung, and they play important roles in tissue remodelling during pulmonary fibrosis [83]. M1 macrophages (‘classically activated macrophages’) contribute to the host defence against pathogens by phagocytosis or by releasing proinflammatory cytokines [84, 85]. So far, M1 macrophages are thought to have both positive and negative roles in fibrogenesis [86–88]. M2 macrophages (‘alternatively activated macrophages’) produce profibrotic mediators such as TGF-β and PDGF [86]. The polarisation of M2 macrophages is influenced by interleukin (IL)-4, IL-13, TGF-β and IL-10, among others. They are implicated in the aberrant wound-healing cascade during fibrosis [89]. We recently reported that the expression of oncostatin M and IL-6 impacts the polarisation of M2 macrophages and the development of bleomycin-induced lung fibrosis [90]. During the progression of IPF, the predominant accumulation of M2 macrophages in fibrotic areas seems to be an important regulator of fibrogenesis [91, 92]. However, newer experimental and human studies suggest that monocytes and macrophages are not only M1 and M2, but can be divided into multiple subsets with functional diversity [93, 94].
The origin of macrophages is the subject of ongoing scientific investigations. Traditionally, it was believed that macrophage populations in adult tissue are continuously replenished by monocytes from the bone marrow [95]. The more contemporary paradigm suggests that tissue-resident macrophages are seeded during embryonic haematopoiesis and self-maintain independently of bone marrow contribution during adulthood [96]. It has also been reported that alveolar macrophages derived from monocytes migrate into the lung during the recovery from inflammation or infection and coexist with tissue-resident macrophages after the resolution of fibrosis [97]. Taken together, these findings suggest that new subtypes of recently discovered profibrotic macrophages may be a posteriori acquired after adulthood. Regardless of their specific origins, it is clear that macrophages contribute to wound repair and fibrosis in a way that goes beyond their inflammatory function and may become promising targets for treatments in the future.
Fibrocytes
Fibrocytes are bone-marrow-derived mesenchymal progenitor cells [98] that express markers of haematopoietic cells (CD34), leukocytes (CD11b, CD13 and CD45) and fibroblast products (collagens I and III and fibronectin) [98, 99]. Although fibrocytes comprise only a small fraction of circulating leukocytes in normal humans, increased numbers of fibrocytes are present in disorders that are characterised by both chronic macrophage-driven inflammation and persistent fibroblast activation [98]. An increase in the percentage of circulating fibrocyte numbers correlates with the abundance of fibroblastic foci in IPF tissue [100, 101]. Our group reported increased percentages of fibrocytes in the peripheral blood of patients with IPF that were predictive of early mortality in these patients [102]. These findings suggest that fibrocytes are involved in the pathogenesis of IPF.
Because isolated fibrocytes have the capacity to differentiate to myofibroblasts in vitro when stimulated by TGF-β [103], the contribution of fibrocytes to the origin of fibroblasts and the progression of fibrosis had been studied for years. However, this differentiation ability has not been directly demonstrated in vivo in wound-healing models with dye-tagged reinfused fibrocytes [98, 103]. Furthermore, fibrocytes transferred into mice did not express α-smooth muscle actin, suggesting fibrocytes may not transform into myofibroblasts in vivo [104]. Collagen production by fibrocytes is significantly lower than fibroblasts [105] and fibrocytes are not an essential source of type I collagen during lung fibrosis [106]. Nevertheless, it is evident that the transfer of fibrocytes promotes pulmonary fibrosis [104, 105] and it is currently believed that fibrocytes contribute to the promotion of fibrosis through effects other than collagen production and differentiation into myofibroblasts, probably through modification of the matrix microenvironment.
Indeed, fibrocytes produce numerous growth factors (e.g. macrophage colony-stimulating factor (M-CSF), TGF-β, FGF, PDGF and VEGF) and chemokines (e.g. IL-8 and macrophage inflammatory protein (MIP)-1α) [98, 107, 108]. Our group also showed that growth factors produced by fibrocytes promote the proliferation of fibroblasts and suggested that the antifibrotic effects of nintedanib are at least partly mediated by suppression of fibrocyte function [108].
Recently, Raghu et al. [109] reported that recombinant pentraxin 2 slowed the decline of lung function in patients with IPF in a phase II study. Biologically, pentraxin 2 inhibits monocyte differentiation into fibrocytes and is also a potent inhibitor of monocyte differentiation into proinflammatory macrophages [109]. Even though fibrocytes may reflect just a small cell population in the fibrotic process, these results are significant because they suggest that therapies targeting fibrocytes and macrophages hold promise and will be investigated in upcoming phase III trials.
Stem cells
Stem cells (e.g. mesenchymal stromal stem cells (MSCs), induced pluripotent stem cells and lung stem cells) have been proposed as a potential therapy for IPF due to their multipotency and role in tissue repair and wound healing. Stem cells produce antifibrotic mediators such as hepatocyte growth factor, FGF-1 and prostaglandin E2 [110–112], and MSCs have been found to elicit a protective effect in mice with bleomycin-induced lung injury [113]. Based on these facts, several clinical trials have been completed to evaluate the safety and efficacy of MSCs in the treatment of IPF [114–116]. These trials concluded that both endobronchial or intravenous administration of stem cells are safe and well tolerated; however, the intervention efficacy of MSCs still needs to be investigated.
ECM abnormality
The lung ECM is constituted of collagens, elastin, glycoproteins, proteoglycans and other components, providing structural scaffolding for cells and mechanical stability of the organ. The ECM also serves as a reservoir for growth factors. In IPF lungs, however, the ECM is extensively modified, which results not only in destruction of lung architecture, but also excessive storage of fibrogenic mediators.
The ECM abnormalities in fibrotic lungs are related to its biomechanical and biochemical properties. ECM stiffness itself contributes to the progression of IPF. The de-cellularised IPF matrix is significantly stiffer than the normal lung matrix [117]. When fibroblasts are cultured on stiff matrices or de-cellularised IPF lungs, they differentiate into activated myofibroblasts characterised by increased α-smooth muscle actin and decreased prostaglandin E2 expression, which are features of IPF myofibroblasts [117–119]. Rho kinase (ROCK) has a role in this phenotypic change by mechanotransduction. In vitro studies showed that ROCK potently stimulates the differentiation of fibroblasts into myofibroblasts [120], and a ROCK inhibitor demonstrated a therapeutic effect in the pulmonary fibrosis model [121]. KD025, a selective ROCK2 inhibitor, is currently in early clinical development (ClinicalTrials.gov identifier: NCT02688647). It seems to be well tolerated and even showed reduced decline of forced lung capacity in a small trial in patients with IPF [122].
Most molecules within the ECM are dynamically turned over and the whole ECM structure is constantly remodelled. In IPF, this remodelling is dysregulated; increased deposition of the individual ECM components partnered with reduced degradation leads to matrix accumulation and fibrosis [117, 123]. ECM fragments such as fibrin, fibronectin and hyaluronan are drastically upregulated in fibrotic ECMs, and have profibrotic effects similar to growth factors [124], suggesting that the compositional changes of the fibrotic ECM alone can drive a profibrotic cell phenotype.
Matrix components have a constant interaction with growth factors, including TGF-β and CTGF. TGF-β is secreted in an inactive form and is bound to latent-associated peptide which prevents interaction with its receptors. Latent-associated peptide is targeted by numerous mediators and proteins including matrix metalloproteinases, which leads to its proteolytic degradation and release of TGF-β. Integrins, especially αVβ6, are known to bind and activate latent TGF-β. Administration of anti-αVβ6 integrin antibodies protects against bleomycin-induced pulmonary fibrosis in mice [125]. A recent proof-of-concept trial with BG00011, a humanised monoclonal antibody against αVβ6 integrin in patients with IPF showed inhibition of phospho-SMAD2 levels in bronchoalveolar lavage cells [126]. These results were encouraging enough to perform a larger study of BG00011 in patients with IPF (ClinicalTrials.gov identifier: NCT03573505).
Mechanical force-induced activation of TGF-β is another important mechanism highlighting the influence of increased tissue and ECM stiffness on cell phenotypes. Recent experiments performed by our group showed that mechanical stretch activates and releases TGF-β in living tissues from fibrotic lungs [127]. This work suggested that even relatively mild mechanical forces, such as distention of the lung tissue during tidal volume breathing, might contribute to progression of IPF through activation of fibrogenic growth factors [128].
Limitations in basic research
In vitro and in vivo models are invaluable for understanding the pathomechanisms in IPF; however, these models have limitations. One example is the difficulty in culturing human primary AECs, which makes in vitro testing challenging. Furthermore, several animal models of IPF, including bleomycin-induced lung fibrosis, fail to fully recapitulate IPF as seen in patients. Despite this, basic science contributes to the field by generating novel in vitro and in vivo models such as induced pluripotent stem cell-generated AECs [129], precision-cut ex vivo lung models [130], lung-on-chip technologies [131] and gene-engineered mice. These systems will be important to appropriately study lung fibrosis and develop effective therapies in the future.
Conclusion
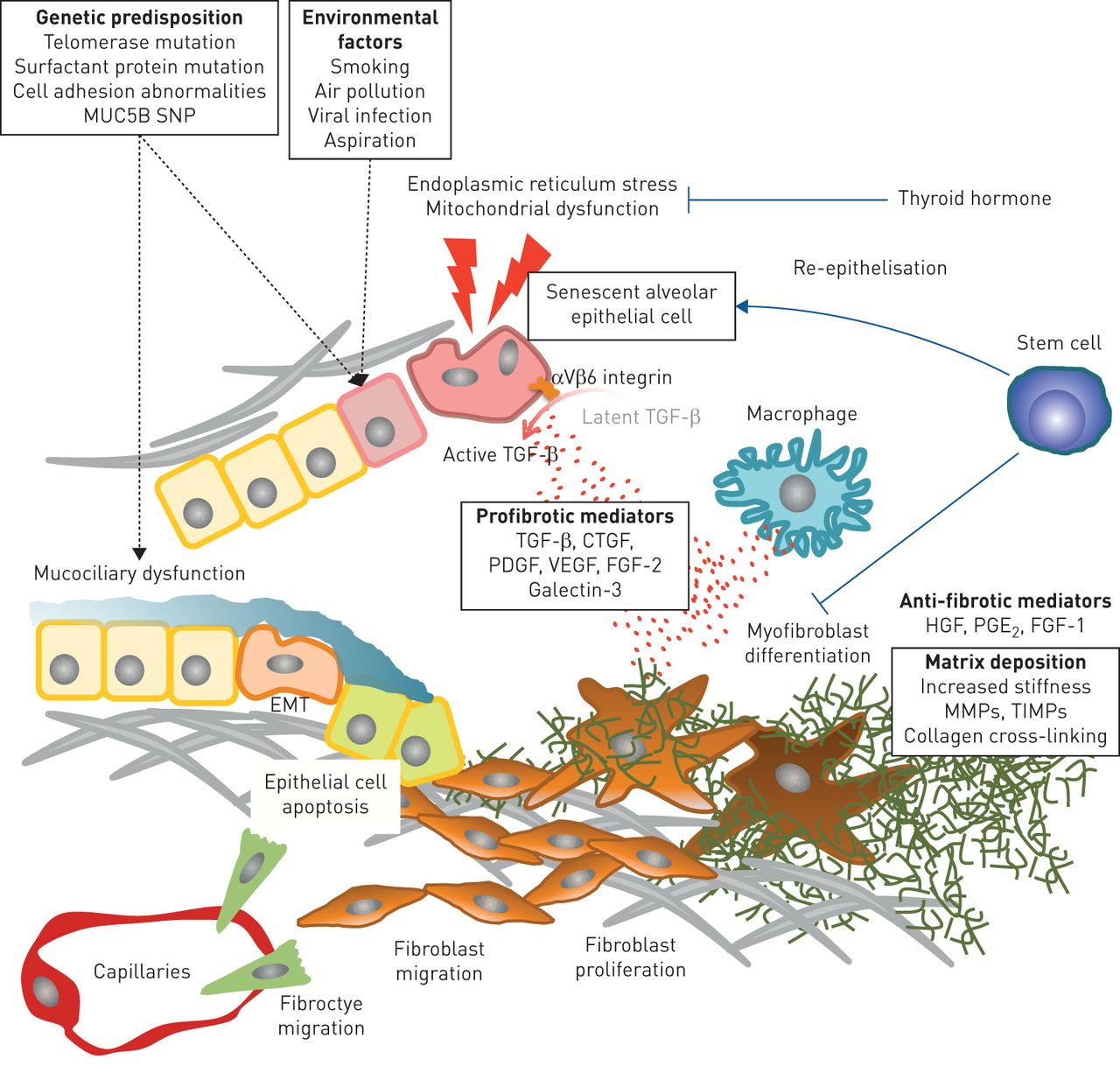
IPF is a complex and progressive lung disorder with limited therapeutic options. Recent advances in the understanding of IPF pathogenesis support the concept that different biological processes are involved sequentially in the development of pulmonary fibrosis (figure 1). The heterogeneity of the disease and the unpredictable clinical behaviour further suggests that these different biological processes are present in one IPF lung at the same time, just in different areas. Chronic epithelial cell damage, most likely caused by as yet unidentified environmental exposures, develop in genetically susceptible individuals. Known genetic mutations or gene variants lead to epithelial cell dysfunction and accelerated ageing. Molecular mediators from dysregulated epithelial cells cause an accumulation of fibrogenic cells and myofibroblast differentiation. These contribute to progression through aberrant ECM deposition. The remodelled lung architecture is composed of a biochemically and biomechanically abnormal matrix, which results in a cyclical loop of progressive fibrosis. Understanding these complex disease steps with their underlying biological basis is key to informing about the best combination of therapies to finally halt the progression of IPF.

{kind=link}
Proposed pathobiological features of idiopathic pulmonary fibrosis. Recurrent epithelial cell injury in genetically susceptible individuals causes senescence of epithelial cells and epithelial mesenchymal transition (EMT), releasing profibrogenic mediators induces fibrocytes/fibroblasts migration and differentiation into profibrotic macrophages/myofibroblasts, resulting in aberrant matrix deposition with destructing lung architecture. SNP: single nucleotide polymorphism; TGF: transforming growth factor; HGF: hepatocyte growth factor; PGE2: prostaglandin E2; FGF-1: fibroblast growth factor-1; FGF-2: fibroblast growth factor-2; CTGF: connective tissue growth factor; PDGF: platelet-derived growth factor; VEGF: vascular endothelial growth factor; MMP: matrix metalloproteinases; TIMP: tissue inhibitors of metalloproteinases.
Footnotes
Conflict of interest: T. Yanagihara reports personal fees from Prometic, outside the submitted work.
Conflict of interest: S. Sato reports grants from Canadian Institute for Health Research, during the conduct of the study.
Conflict of interest: C. Upagupta reports grants from Canadian Institute for Health Research, during the conduct of the study.
Conflict of interest: M. Kolb reports grants from Canadian Institute for Health Research, during the conduct of the study; grants and personal fees from Roche, Boehringer Ingelheim and Prometic, grants from Actelion, Respivert and Alkermes, and personal fees from Genoa, Indalo and Third Pole, outside the submitted work.
Support statement: This study was funded by the Canadian Institutes of Health Research, Institute of Circulatory and Respiratory Health (MOP136950). Funding information for this article has been deposited with the Crossref Funder Registry.
- Received March 14, 2019.
- Accepted May 11, 2019.
- Copyright ©ERS 2019.
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










