





Abstract
Pulmonary arterial hypertension (PAH) is a condition characterised by increased pulmonary vascular resistance which can lead to right heart failure and premature death. It imposes a significant burden on patients' lives, affecting their physical, emotional and social wellbeing. Pharmacological therapies are the mainstay of treatment; while they are not curative, they can alleviate patient suffering, improve quality of life and delay disease progression. Despite these therapies, disease progresses in a significant number of patients, who are faced with the debilitating symptoms of PAH and treatment adverse effects. Palliative care is focused on providing relief from symptoms caused by a chronic illness. Palliative care aims to improve the health-related quality of life for patients and families, and although it is deemed appropriate at any stage of disease, it is most helpful when explored early in the course of disease. Importantly, palliative care can be provided in concert with pharmacological treatment. Despite its potential benefits, palliative care is frequently underutilised. There is a paucity of clinical studies testing the impact of palliative care in PAH which prompted us to summarise the available evidence, recognise obstacles in its utilisation and identify areas for future research.
Abstract
Palliative care remains an underutilised treatment option in PAH. This article attempts to highlight its importance in the management of PAH and highlight the barriers faced by both physicians and patients in seeking palliative care. http://ow.ly/quBH30mt9Pl
Introduction
Pulmonary arterial hypertension (PAH) is a complex, progressive disease with poor long-term survival [1]. The prognosis of PAH has improved in the past two decades, in part due to the approval of several PAH-specific therapies. However, the Registry to Evaluate Early and Long-term PAH Disease Management (REVEAL) showed a 50% mortality at 7 years [2]. In addition to the high mortality, the morbidity of the disease profoundly affects the health-related quality of life (HRQoL) of these patients [3, 4].
Pharmacological therapies are essential in the management of patients with PAH. While some PAH-specific medications delay disease progression, they are not curative. In fact, in a good proportion of patients the disease will progress, eventually leading to right heart failure and death [1]. In addition, PAH-specific medications cause adverse effects that further affect the patients' HRQoL [5].
Physical and emotional symptoms palliation is important to improve the HRQoL of patients living with chronic diseases like PAH. However, palliative care interventions are often overlooked by clinicians since they are associated with end-of-life care. This fact results in the underutilisation of palliative measures [6]. Furthermore, chronic diseases not only affect the HRQoL of patients, but also their caregivers and family members. Palliative care is a method of improving the HRQoL of both patients and caregivers when faced with incurable diseases. This entails timely intervention to 1) alleviate suffering by decreasing symptoms such as dyspnoea and pain and 2) provide a system of support oriented towards psychosocial, emotional and spiritual wellbeing [7, 8].
In spite of abundant data on the treatment of PAH [9], there is a paucity of clinical studies on the role of palliative care in the management of PAH. The important clinical implications prompted us to summarise the available evidence on the use of palliative care in patients with PAH, recognise obstacles in its utilisation and identify areas for future research.
Health-related quality of life in PAH
Pulmonary arterial hypertension imposes a significant burden on patients' lives in many aspects, with studies showing poor HRQoL indices [3, 4, 10, 11], that might be as severe as those found in other serious and debilitating conditions such as cancer [12], interstitial lung disease [13] and spinal cord injury [14]. Pharmacological therapies for PAH alleviate patient suffering and improve HRQoL [15–20]; however, treatment-related adverse effects and parenteral routes of administration can negatively influence HRQoL [21, 22].
The overall HRQoL is measured by assessing a variety of indicators, including functional status, physical, emotional, social and spiritual wellbeing. The Pulmonary Arterial Hypertension – Symptoms and Impact (PAH-SYMPACT) questionnaire assesses disease-specific patient-reported outcomes and has recently been validated for use in PAH patients [23, 24]. This questionnaire assesses the symptoms of PAH in the previous 24 h and the impact PAH had on patients' lives in the preceding 7 days. Table 1 shows various studies that assessed HRQoL in PAH patients and the tools used to assess it.
Studies assessing the quality of life (QoL) in patients with pulmonary arterial hypertension (PAH)
PAH significantly affects the physical capabilities of patients. Symptoms of shortness of breath, fatigue and exhaustion remarkably limit patients' ability to execute activities of daily living [25]. This reduced physical activity negatively impacts HRQoL and survival [4, 26]. In addition, patients with PAH have significant psychological morbidity. In fact, the diagnosis of PAH creates emotional and psychological distress, reflected as feelings of frustration, anger, low self-esteem and worthlessness [25, 27]. Indeed, patients with worse functional class and reduced exercise capacity often have a higher prevalence of depression [10]. An observational study by McCollister et al. [10] showed that 55% of patients with PAH have depression and 15% have major depressive illness. In another study, Löwe et al. [28] found that 35% of patients with PAH suffer from psychological disorders, with the most common being depression, closely followed by panic attacks; conditions that are rarely treated. The authors found a higher prevalence of psychological symptoms in those with worse functional class. Shafazand et al. [3] reported anxiety and depression in 21% and 8% of patients with PAH, respectively. Given the high prevalence of these psychological conditions in this patient population, clinicians should be cognisant about these conditions and assess their patients during clinical encounters to prevent under-recognition and under-treatment.
Anxiety is augmented by the patients' limited knowledge regarding PAH, including the uncertainties regarding treatment and prognosis. Many patients and caregivers resort to finding information on the internet. However, the information readily available on the web might not be current, totally accurate or applicable to the patient's specific condition. Moreover, given that PAH is a rare condition, primary care physicians might lack adequate knowledge and/or experience to educate confidently and put patients at ease [29]. Some patients cope with these uncertainties by making memories with their loved ones and finding humour in their present setting [5].
Unexpected changes in the patients' and caregivers' lives contribute to the overall suffering in PAH. Worsening HRQoL in patients with PAH is associated with reduced social activity and emotional wellbeing [11]. Patients frequently face a loss of financial security and social status, as they become unable to maintain employment [10], travel obligations or social activities [25, 27]. In addition, marital relationships can suffer as sexual intimacy diminishes given physical limitations, reduced self-esteem and a partner's fear of affecting the patient's condition [25].
The burden of disease is not limited to patients, but also extends to caregivers. In surveys, caregivers voiced feelings of fear about losing their loved ones, as well as symptoms of stress and exhaustion as a result of the increased responsibilities of caring for them [25]. Often, the caregivers' employment and social life is affected, contributing a decrease in the household income and overall emotional support [25]. A cross-sectional study by Hwang et al. [30] showed that 14% of caregivers of patients with PAH had to quit their jobs or reduce work hours to help provide care. The same study showed that 14% of caregivers had a Patient Health Questionnaire-8 score ≥10, suggestive of clinical depression [30].
As the disease progresses, the magnitude of physical symptoms including dyspnoea, chest pain and fatigue increases [31]. With more severe symptoms, the functional class of patients declines and survival worsens [31–33]. In addition, the progression of physical symptoms is often accompanied by proportional worsening of psychological suffering and emotional distress experienced by patients and their caregivers [10, 25]. The psychological and emotional distress contributes to further decline in HRQoL. This highlights the importance of a multimodality approach to help those patients and their caregivers ease their sufferings and improve their HRQoL.
Palliative care in PAH
The Center to Advance Palliative Care defines palliative care as a specialised medical care for people living with serious illnesses. Palliative care is focused on providing relief from symptoms and stress caused by any chronic illness, focusing in improving the HRQoL of the patient and family. Palliative care is provided by a team of doctors, nurses and other specialists who work with the patient's physicians to provide an extra layer of support. Palliative care is appropriate at any age or stage of disease and importantly, it must be provided in concert with curative treatment.
Saunders [34], defines “total pain” as the integration of physical and emotional suffering; and by addressing it as a whole (physical symptoms, mental distress, social problems and emotional difficulties), the HRQoL of patients and caregivers can be improved. Since patients with PAH have a noticeable degree of “total pain”, it is logical that a palliative medicine intervention might be beneficial. Nevertheless, Fenstad et al. [6] reported on the barriers to receiving palliative medicine, including a lack of referral and the misperception that palliative care is equivalent to losing hope or is limited to end-of-life care [6, 11].
Many patients and caregivers do not know the role of palliative care, or perceive that is similar to hospice treatment [29]. A common misconception is that patients can no longer be treated for PAH when receiving palliative care [11]. Furthermore, Fenstad et al. [6] noted that some physicians erroneously think that a palliative care referral is not possible while patients receive i.v. prostanoids. Although physicians are generally comfortable in discussing end-of-life options, not many are at ease when addressing HRQoL issues [6]. Physicians commonly ask about pain, but not all are comfortable giving pain medications or neuromodulator agents. In fact, only 14% of physicians feel comfortable in managing depression [6]. It is unclear how these findings apply to symptoms like anxiety, nausea, poor appetite and fatigue [6, 35].
Adverse effects of PAH-specific therapies deteriorate patients' HRQoL. PAH medications can cause significant adverse effects such as headache, nausea, diarrhoea, flushing, swelling, etc. These adverse effects may limit patients' daily activities and social plans, contributing to depression or loneliness. Furthermore, the route of medication administration may contribute to patients' distress. For instance, the subcutaneous delivery of treprostinil is frequently associated with site pain [36]. Additionally, i.v. delivery methods require permanent i.v. access, which may be associated with infections and catheter displacement or malfunction.
Palliative care in PAH versus other serious illnesses
In some aspects, PAH behaves like certain cancers, since PAH is a progressive disease which may lead to premature death despite the best available treatments. Some cancers, if diagnosed early, can be treated with curative intent; however, there are no curative treatments for PAH irrespective of the stage of diagnosis. Palliative care plays a significant role in cancer management, but less so in PAH. A randomised clinical trial of palliative care interventions in lung and gastrointestinal malignancies showed that early involvement of a palliative care teams improved patient and caregiver experiences [37]. Although these findings may hold true in PAH, there are still no data to support this belief.
Studies have shown that one of the barriers to referring patients with progressive lung disease for palliative medicine is the unpredictability of the disease [38]. Unlike malignancy, physicians find it hard to predict nearness of death with nonmalignant diseases that have exacerbations that alternate with periods of better control of the disease [39, 40]. When prognostic uncertainty is coupled with a cloudy understanding of the differences between palliative and hospice care, opportunities to improve patients' HRQoL are lost [41]. Several predictors of disease severity and survival have proven to be valid in PAH, including World Health Organization (WHO) functional class, 6-min walk test (6MWT), N-terminal pro-brain natriuretic peptide (NT-proBNP), echocardiographic measures of right ventricular function and haemodynamic determinations. In addition, there are various risk assessment tools that use a combination of these measurements [9, 31–33]. These predictors of disease severity and survival help clinicians assess the prognosis and disease severity, diminishing the disease unpredictability faced in other conditions.
Palliative therapies
Palliative care in PAH can be broadly categorised into invasive and non-invasive. The invasive modalities of palliative care include atrial septostomy [42], right ventricular assist devices [43] and pulmonary artery denervation [44]. The non-invasive interventions include educating patients about their condition, providing financial assistance and information on health insurance coverage, encouraging patients to join support groups, offering spiritual support and screening for depression and providing counselling if needed [5, 10, 30]. Figure 1 summarises palliative interventions in patients with end-stage PAH.

Overview of palliative care in patients with end-stage pulmonary artery hypertension.
Invasive palliative therapies
Atrial septostomy is a percutaneous procedure, by which a right-to-left shunt is created in the atrial septum, allowing decompression of the right side of the heart [45]. Several studies have shown that in the right candidates, atrial septostomy may improve symptoms, haemodynamics and potentially survival in patients with PAH [46–50] (table 2). Sandoval et al. [42] reported an improvement in functional class, 6-min walking distance (6MWD) and haemodynamic parameters (cardiac index and right ventricular pressure) following atrial septostomy in PAH patients. In another retrospective analysis, Chiu et al. [46] described an improvement in symptomatic heart failure and occurrence of syncope after atrial septostomy, without significant differences in haemodynamic determinations.
Outcomes of various palliative interventions in patients with pulmonary arterial hypertension (PAH)
Another intervention is the placement of a right ventricular assist device (RVAD). Punnoose et al. [43] modelled the cardiovascular system in simulated cases of PAH and evaluated the effects of RVAD flow rate on various haemodynamic measures. With increasing RVAD speed, there was an increase in total pulmonary blood flow, mean pulmonary artery pressure, diastolic pulmonary artery pressure, left ventricular filling pressure and cardiac output, as well as a decrease in right atrial pressure. However, this theoretical model had limitations since it reflected only the acute effect of RVAD on haemodynamics, assuming fixed pulmonary vascular resistance ventricular interactions.
Pulmonary artery denervation (PADN) involves radiofrequency ablation of sympathetic nerve fibres located at the level of the main pulmonary artery bifurcation. Chen et al. [44] used this methodology for the treatment of idiopathic PAH not responding to medical therapy. Compared to the control group, those patients with idiopathic PAH who underwent PADN had a significant reduction in mean pulmonary pressure (from 55±5 mmHg to 36±5 mmHg; p<0.01) and pulmonary vascular resistance (from 1883 dyn·s·cm−5 to 763 dyn·s·cm−5), an increase in cardiac output, improvement in the 6MWD (from 324±21 m to 491±38 m; p<0.006), Borg dyspnoea index and WHO functional class (from 3.6 to 1.6) at 3 months following the procedure. This improvement in haemodynamics and functional capacity shows that PADN might be a promising palliative procedure that needs further investigation.
In summary, the role of these invasive palliative interventions in PAH needs to be further investigated. While PADN have been shown to improve symptoms, functional class and haemodynamics in PAH patients [44], its effect in survival has not been studied and there are limited data to provide treatment recommendations. The use of RVAD in PAH also needs further investigation to better define its role [43]. Atrial septostomy has been performed as a palliative intervention in PAH in patients with end-stage PAH and sometimes as a bridge to transplant. Atrial septostomy was associated with improvement in symptoms, haemodynamics, functional class and potentially survival [42, 46, 49, 50]. However, these recommendations are based on anecdotal evidence, which again emphasises the need for further studies.
Non-invasive palliative therapies
Patients with PAH and their families experience physical, emotional, social and spiritual distress. The degree of physical and emotional distress is comparable or even more pronounced than patients living with cancer [35]. Interestingly, patients with PAH rate pain scores similar to patients with cancer, and symptoms of nausea almost four times higher than cancer patients [35]. With both opioid- and non-opioid-based therapies, palliative medicine physicians can work with patients towards the goal of living with less pain. Nausea can be controlled or eradicated with medications from a variety of drug classes. Fatigue and anorexia may be improved with medications to boost energy and appetite. Anxiety and depression are alleviated with medications and counselling. Counselling helps patients develop coping skills and reach peace with their “new state”. Addressing each physical symptom contributes to the patient's global sense of wellbeing; with the goal of living with no or controlled pain or nausea, as well as higher energy and appetite, improvements that can help boost spirits and regain emotional stability. In addition, caregivers and families feel relief when the patient's suffering is eased.
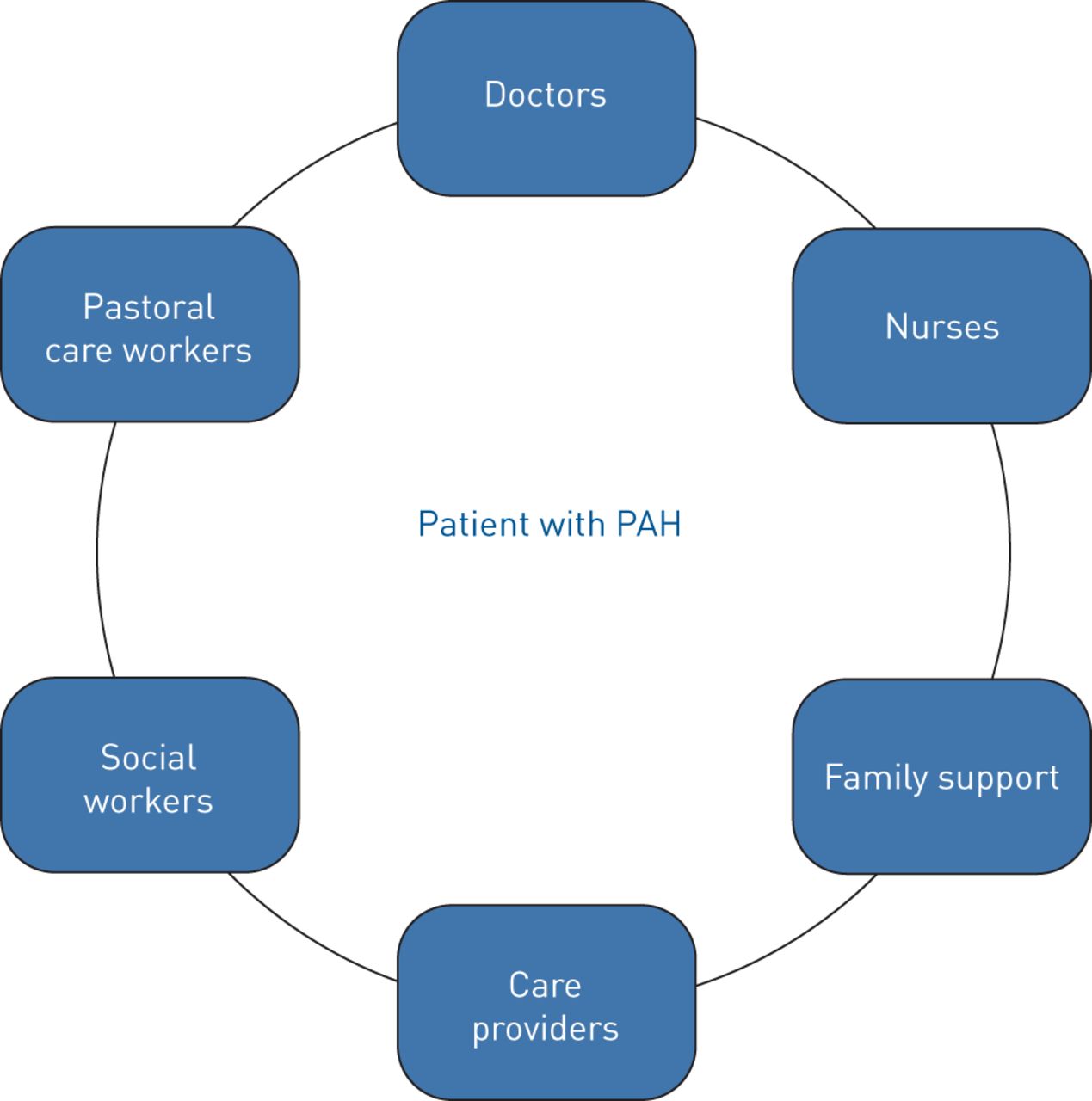
Social workers are part of every palliative care team. Social workers offer supportive listening and counsel patients and families, helping navigate changes in social status and concerns about the future. Social workers can help families with insurance and financial issues, which can seem overwhelming as family income and roles evolve as the patient's disease progresses. Another member on the palliative care team is a pastoral counsellor. Patients with a life-limiting disease and their families struggle with questions of faith, the meaning of life and fear of the unknown. Access to a pastoral counsellor can provide a space for discussing these spiritual issues. Figure 2 shows components of the palliative care team.

{kind=link}
{kind=link}
Structure of the palliative care team. PAH: pulmonary arterial hypertension.
While receiving palliative care, patients may receive parenteral PAH therapies under the care of their pulmonary hypertension physician. This can be at home, with home palliative medicine services until the patient reaches the point at which symptoms cannot be adequately controlled. Patients at the end of life may receive care in a hospital setting with careful consideration of patients' and families' expectations and goals. Tonelli et al. [1] showed that fewer than half the PAH patients who died had advance healthcare directives, and most (80%) of these deaths occurred in a healthcare setting, predominantly in the intensive care unit (52%) [1]. These results emphasise the need for a more proactive approach in discussing of end-of-life care and making sure that patients with this progressive disease have time to consider options for end-of-life care. This approach helps relieve the stress and anxiety of family members who may have to make important health decisions if the patient becomes incapable of doing so.
Palliative care and lung transplant
Referral for lung transplant in PAH is recommended for patients with New York Heart Association functional class of III or IV despite maximal medical therapy, patients on parenteral prostanoids and patients with rapidly progressive disease, as evidenced by worsening 6MWT, NT-proBNP, right ventricular function and haemodynamics [51]. Patients with PAH whose mortality risk is high (estimated 1-year mortality >10%) based on comprehensive assessment of multiple variables (clinical signs of right heart failure, syncope, WHO functional class, 6MWD, NT-proBNP, cardiopulmonary exercise testing, echocardiography and haemodynamics) should be considered for lung transplantation [9]. Given the symptom burden and the sometimes prolonged waiting time for lung transplant, concomitant palliative care services are crucial to help improve HRQoL and minimise the suffering of PAH patients.
Palliative care for patients on the lung transplant waiting list involves taking care of symptoms, planning for advance care and providing emotional and spiritual support [52]. A retrospective cohort study of lung transplant candidates referred for palliative care by Colman et al. [52] showed that palliative care could be provided without harming the chances of a successful transplant. Palliative care should not be delayed until the moment when a patient is disqualified for transplantation. Pulmonary hypertension referral centre clinics should manage these end-stage patients in multidisciplinary clinics where they can get support from social workers, pastoral counsellors and palliative care physicians.
Because PAH is a progressive and life-limiting disease, palliative care involvement at an early stage in the care plan is very important, it provides an “extra layer of support” offered by a multidisciplinary palliative care team. It is important that physicians come to understand that palliative care is not the same as end-of-life care, and that palliative care is best when offered alongside life-prolonging therapies [53]. As there is a better understanding of what palliative care can offer, patients should know that a referral to palliative care is not “giving up,” but instead is an effort to thoroughly investigate all possible avenues for making quality of life the best it can be.
Conclusion
Palliative care has much to offer to patients with PAH, whose suffering extends beyond the physical realm into the psychological, spiritual and social domains. Palliative care improves functional status, exercise tolerance, haemodynamic values and viability to receive transplants. We need to better understand the reasons for the underutilisation of palliative care interventions and educate patients, caregivers and physicians on this treatment approach in order to help alleviate the suffering of patients with PAH.
Footnotes
Provenance: Submitted article, peer reviewed.
Author contributions: G. Khirfan participated in literature review, collection of relevant information, writing and critical revision of the manuscript for important intellectual content and final approval of the manuscript submitted. A.R. Tonelli participated in writing and critical revision of the manuscript for important intellectual content and final approval of the manuscript submitted. J. Ramsey participated in writing and critical revision of the manuscript for important intellectual content and final approval of the manuscript submitted. S. Sahay participated in concept designing, writing and critical revision of the manuscript for important intellectual content and final approval of the manuscript submitted.
Conflict of interest: G. Khirfan has nothing to disclose.
Conflict of interest: A.R. Tonelli has nothing to disclose.
Conflict of interest: J. Ramsey nothing to disclose.
Conflict of interest: S. Sahay has received a grant from the American College of Chest Physicians (PAH research award) and is a speaker on a panel for United Therapeutics, Actelion and Bayer pharmaceuticals.
- Received August 7, 2018.
- Accepted October 16, 2018.
- Copyright ©ERS 2018.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










