





Abstract
Neurofibromatosis type 1 (NF1), also known as von Recklinghausen disease, is a frequent autosomal dominant genetic disorder with a prevalence of 1 in 3000. Pulmonary hypertension (PH) associated with NF1 (PH-NF1) is a rare but severe complication of NF1 and is classified as Group 5 PH, defined as “PH with unclear and/or multifactorial mechanisms”. A literature review in PubMed on the association between NF1 and PH identified 18 articles describing 31 cases. PH-NF1 was characterised by a female predominance, an advanced age at diagnosis, an association with parenchymal lung disease in two out of three cases and poor long-term prognosis. NF1 is generally associated with interstitial lung disease but some cases of severe PH without parenchymal lung disease suggest that there could be a specific pulmonary vascular disease. There is no data available on the efficacy of specific pulmonary arterial hypertension treatment in PH-NF1. Therefore, these patients should be evaluated in expert PH centres and referred for lung transplantation at an early stage. As these patients have an increased risk of malignancy, careful assessment of the post-transplant malignancy risk prior to listing for transplantation is necessary. Clinical trials are needed to evaluate promising treatments targeting the RAS-downstream signalling pathways.
Abstract
Pulmonary hypertension is a rare but severe complication of neurofibromatosis type 1. There are no data about the efficacy of specific PAH treatment in this disease and lung transplantation should be discussed at an early stage. http://ow.ly/JMU030lezfY
Introduction
Neurofibromatosis type 1 (NF1), also known as von Recklinghausen disease, is a frequent autosomal dominant disorder which can require the respiratory physician's expertise. Indeed, NF1 is a multisystem disease and lung disease, especially pulmonary hypertension (PH), can be one of the most severe complications. PH associated with NF1 (PH-NF1) is classified as Group 5 PH, defined as “PH with unclear and/or multifactorial mechanisms” (table 1) [1]. This is a heterogeneous group including several disorders with multiple mechanisms, for which there is neither data nor recommendations regarding the use of pulmonary arterial hypertension (PAH) approved drugs. The only treatments available target the underlying disease. An improved understanding of PH-NF1 should help advance the pathophysiology of PH in general. This article is about NF1, however, it is interesting to note that neurofibromatosis type 2 is a different disease, caused by a different genetic mutation, and that to our knowledge, there is no described case of PH in neurofibromatosis type 2.
Classification of group 5 pulmonary hypertension according to European Society for Cardiology/European Respiratory Society guidelines
Clinical presentation of NF1
NF1 is characterised by multiple café au lait spots which occur in 95% of patients, axillary and inguinal freckling in 70% of patients, several discrete benign neurofibromas within the dermis in 95% of patients, and also iris Lisch nodules in 95% of patients [2]. Less common but potentially more severe manifestations can also occur, in particular with a predisposition for tumours. The most frequent tumours are nodular neurofibromas which occur in peripheral nerves and can grow to an enormous size, as well as plexiform neurofibromas which are usually congenital and occur in 30% of patients. These tumours are an important cause of morbidity as they affect long portions of nerves, can infiltrate the nerve and surrounding tissue and in ∼2–16% of patients transform to malignant peripheral nerve sheath tumours. Optic and other central nervous system gliomas occur in 15% of patients with potentially severe complications as they produce symptoms in 2–5% of cases. Phaeochromocytoma is rarely associated with the disorder, affecting between 0.1 to 5.7% of patients [3]. Intestinal tumours like carcinoids are also more frequent than the general population, and there is a higher incidence of malignancy in general in NF1.
NF1 is associated with osseous lesions like scoliosis, dysplasia of the sphenoid wing and thinning of long bones. Hypertension is present in 6% of patients and can be caused by renovascular disease, coarctation of the aorta and phaeochromocytoma. Learning disabilities are present in at least 50% of patients [4] and there is also an increased risk of epilepsy and headaches.
Cardiac abnormalities like secundum atrial septal defect, ventricular septal defect, mitral or aortic insufficiency, hypertrophic cardiomyopathy, and intracardiac tumours have also been described and are severe complications of NF1 [5, 6].
Finally vascular lesions are less frequently reported but are some of the most severe complications of NF1 [7, 8]. These include occlusive or aneurysmal arterial lesions, arterio-venous malformations, coarctation or segmental hypoplasia of the abdominal aorta with or without renal artery ostial stenosis (which can cause renovascular hypertension), occlusive coronary artery disease, visceral vasculopathy causing ischaemic bowel disease and retroperitoneal or abdominal bleeding, and peripheral vascular disease [7].
The diagnosis of NF1 is clinical, based on criteria proposed by the National Institute of Health (NIH) Consensus Development Conference [9] (table 2). If two or more of the seven criteria of the NIH conference are present in the same patient, the diagnosis is established. Diagnosis by genetic testing is possible but is usually not required because of the typical clinical features of the disease and of the great variety of mutations of the neurofibromin 1 (NF1) gene.
Diagnostic criteria for neurofibromatosis type 1 (NF1) [9]
NF1 is fully penetrant in adults, but many manifestations of the disease increase in frequency or severity with age [10]. The disease features are extremely varied, even within the same family. Most studies have not found an evident relationship between particular NF1 mutations and the resulting clinical phenotype. The average life expectancy of patients with NF1 is reduced by 10–15 years and cancer is the most common cause of death [11, 12]. Disease treatment requires multidisciplinary life-long follow-up adapted to the patient's age [13]. It includes referral to specialists for treatment of complications. Surgery to remove both benign and malignant tumours or to correct skeletal manifestations is sometimes warranted, as well as arterial reconstruction, excision of arterio-venous malformations, clipping or embolisation of vascular lesions [8]. Annual physical examination by a physician familiar with the disorder is recommended. Other recommendations include ophthalmological examinations annually in children and less frequently in adults, regular developmental assessment in children, regular blood pressure monitoring, and magnetic resonance imaging for follow-up of clinically suspected intracranial and other internal tumours. Regular pulmonary examination, chest imaging and echocardiographic follow-up are also warranted because lung and cardiovascular complications are now well-documented complications of NF1.
Genetics, biology and pathophysiology in NF1
NF1 is a frequent autosomal dominant genetic disorder with a prevalence of 1 in 3000 [10, 14] and near-complete penetrance before the age of 5 years. The disease is caused by mutations of the NF1 gene, identified in 1990, which is located at chromosome 17q11.2 [11] and comprises 60 exons. It encodes a cytoplasmic protein named neurofibromin, which holds 2818 amino acids and has a role in tumour suppression [2]. Indeed, neurofibromin has a guanosine triphosphatase (GTPase)-activating protein domain that is responsible for decreasing the level of Ras bound to guanosine triphosphate (GTP) by hydrolysing GTP bound to small monomeric GTP-bound Ras [15]. This GTPase activity acts as a negative regulator of signal transmitted by Ras [16] and its loss is associated with the activation of several transcription pathways: the mitogen-activated protein kinase (MAPK) pathway ending by ERK activation [17] and also the mammalian target of rapamycin (mTOR) pathway, mediated by an activation of the PI3kinase-AKT pathway [18, 19] and by the tuberous sclerosis protein 1–tuberous sclerosis protein 2 complex (figure 1) [20].

Schematic representation of the downstream signalling pathways of neurofibromin. Neurofibromin inhibits the activity of Ras by its guanosine triphosphatase activity. Loss of activity of neurofibromin in neurofibromatosis type 1 (NF1) leads to the activation of different pathways mediated by RAS, namely the mitogen-activated protein kinase cascade leading to activation of ERK and mammalian target of rapamycin (mTOR) pathway. These pathways activate nuclear transcription factors and are responsible for endothelial cell differentiation, proliferation, increased survival and migration. PI3K: phosphatidylinositol 3-kinase; Rheb: Ras homolog enriched in brain; TSC: tuberous sclerosis protein.
Inactivation of the gene through mutation leads to a loss of neurofibromin and to a constitutive activation of these pathways. It leads to the deregulation of cell proliferation and differentiation and to the development of benign neurofibroma-like tumours and malignant peripheral nerve sheath tumours, and probably also to the lung complications seen in NF1. About half of all cases result from neomutations [10]. A wide variety of mutations in the NF1 gene have been found in NF1 patients, but no recurring mutation has been identified.
Pulmonary hypertension
A PubMed literature review of the association between NF1 and PH found 18 articles describing 31 cases of pre-capillary PH (defined by a mean pulmonary artery pressure (mPpa) ≥25 mmHg and pulmonary artery wedge pressure <15 mmHg) measured by right heart catheterisation (table 3) [21–38]. None of these patients had identified risk factors for PAH, including anorexigen use, conditions associated with PAH (portal hypertension, HIV infection, connective tissue diseases, congenital heart disease) or chronic thromboembolic disease.
Published articles on pulmonary hypertension associated with neurofibromatosis type 1
Comparison of key distinguishing features between pulmonary arterial hypertension (PAH) and pulmonary hypertension associated with neurofibromatosis type 1 (PH-NF1)
Although NF1 affects male and female patients without sex predominance, there were 25 females but only six males (male:female sex ratio was 1:4.2), suggesting a female predominance in PH-NF1, as is generally seen in idiopathic and heritable PAH [39, 40].
It is interesting to note that PH occurred late in the course of NF1, with a median (range) age at diagnosis of 57 (50–65) years, in contrast with heritable PAH, which is characterised by a younger age at diagnosis (mean age 35.7 years in BMPR2 mutation carriers and 21.8 years in ACVRL1 mutation carriers), or with patients with idiopathic PAH (without an identified mutation, mean age 47.6 years) [41]. Dyspnoea and signs of right heart failure were the principal symptoms leading to evaluation for associated PH. Most patients had severe haemodynamic impairment at diagnosis, with a low cardiac index (median (range), 2.3 (2.0–2.7) L·min−1·m−2) and high levels of mPpa (median (range) 49 (39–60) mmHg) and pulmonary vascular resistance (median (range) 14.2 (8.9–19.0) Wood unit). None of the patients had a vasodilator response to nitric oxide. Acute vasodilator response with nitric oxide is reported in about 10% of idiopathic PAH, and is associated with long-term response to calcium channel blockers and an excellent prognosis [42]. However, it has been shown that the proportion of acute vasodilator responders was low in patients with heritable PAH [41], as well as in PAH associated with other conditions [43].
Moreover, most of the patients had severe exercise limitation with 75% of the patients in New York Heart Association functional class III or IV at diagnosis and a median (range) 6-min walk distance of 230 (153–300) m. As described later in this article, NF1 may be associated with parenchymal lung involvement. In the 31 reported cases of PH-NF1, lung involvement was reported in 22 patients and was in accordance with data reported by Zamora et al. [44]. Lung involvement manifested itself by mosaic perfusion with ground-glass opacities (n=10), lung cysts (n=10), interstitial septal infiltrates (n=5), large bullae (n=2), mediastinal schwannomas (n=2), pneumothorax (n=1), mild emphysema (n=1), intra-thoracic meningocele (n=1), lung nodules (n=1) and a suspicious lung mass (n=1). It could be suggested that precapillary PH observed in these patients may be due to vascular rarefaction and hypoxic vasoconstriction associated with parenchymal lung disease. However, nine (29%) out of 31 patients with confirmed severe PH had no significant lung involvement on high-resolution computed tomography (HRCT) of the chest and had normal pulmonary functional tests (PFTs). Furthermore, in most patients, the median (range) spirometry and lung volume measurements were in the normal range: forced expiratory volume in 1 s was 93 (71–104) %, forced vital capacity was 83 (72–99) % and total lung capacity was 94 (78–103) %. However, diffusing capacity of the lung for carbon monoxide was importantly decreased in most patients (median (range) 48 (27–57) %) suggesting significant pulmonary capillary involvement.
In this article, we found that most patients received conventional therapy for PH including oxygen and diuretics if needed and sometimes anticoagulation. In addition, despite the absence of recommendations, 22 patients received specific PAH therapies, including endothelin receptor antagonists (n=17), phosphodiesterase type-5 inhibitors (n=16), and i.v. prostanoids (n=13). Four patients also received calcium antagonists although they had no acute vasodilator response with nitric oxide. Two patients had balloon atrial septostomy and only one had lung transplantation. Another patient with PH predominantly due to restriction from an intrathoracic meningocele and scoliosis was successfully treated by noninvasive ventilation alone [38].
The outcomes of these patients reported in the literature were characterised by a limited response to specific PAH therapy and poor outcomes. Indeed, 13 out of 31 patients died with a median (range) delay of 24 (5–39) months. Of note, one patient was treated with the tyrosine kinase inhibitor (TKI) sorafenib and experienced mild clinical and haemodynamic improvement after 3 months; however, no data on long-term response was available [31]. Nevertheless, the benefit/risk ratio of sorafenib in PH is debatable due to possible impairment of cardiac output [45] and imatinib, another TKI tested in a PAH clinical trial, was associated with an increased incidence of subdural haemorrhage [46, 47]. Furthermore, it has been demonstrated that the second-generation TKI dasatinib may induce pulmonary endothelial dysfunction and severe PAH [48–50]. Another patient developed pulmonary oedema with specific PAH treatment and died 3 months later [35]. An autopsy confirmed the diagnosis of capillary haemangiomatosis, a condition similar to pulmonary veno-occlusive disease, known to have a bad response to specific PAH treatment and a poor prognosis [51]. Pulmonary veno-occlusive disease has also been suspected in another case with respiratory failure and death a short time after beginning a specific PH-treatment [32]. This limited response to specific PAH therapies and the poor prognosis of PH-NF1 emphasise the importance of referral for lung transplant assessment early in the course of the disease in eligible patients. While evaluating a patient with NF1 for lung transplantation, one must keep in mind that these patients have an increased risk of cancers and that the immunosuppression required for transplantation increases this risk. Among the 31 reported cases of PH-NF1, one had lung transplantation and was making satisfactory progress after 8 months [28]. Merlo et al. [52] described two other cases of lung transplantation in patients with NF1 because of advanced tobacco-induced chronic obstructive pulmonary disease but without PH. One patient was doing well after 5 years but the second patient developed post-transplant lymphoproliferative disorder and a massive intra-abdominal sarcoma consistent with a malignant nerve sheath tumour 9 months after lung transplantation and died 2 months later [52]. This emphasises the risk of immunosuppressive treatment in these patients and the attentive follow-up needed after lung transplantation.
Four patients had vascular histologic assessment: one pulmonary tissue biopsy [27], one after lung transplantation [28] and two after autopsy [23, 35]. The presence of vascular remodelling of the small pulmonary arteries with intimal and medial thickening and fibrosis was the common finding in these samples. Montani et al. [28] showed that the pulmonary vascular involvement included arterial remodelling with eccentric thickening of the intimal layer and uniform wall thickening through hyperplasia of pericytes/smooth muscle cells in most of the small pulmonary arterioles. Plexiform lesions were also found [23], along with areas of alveolar capillary engorgement and tortuosity with a diagnosis of pulmonary capillary haemangiomatosis [35] and lung interstitial fibrosis with partial loss of parenchymal architecture [28]. These observations reinforce the hypothesis of a disproportionate pulmonary vascular involvement in the case of interstitial lung disease in NF1.
The NF1 gene was screened in nine patients and heterozygous germline mutations of the NF1 gene were identified in all cases. NF1 mutations were of different types, including short deletions, nonsense and missense mutations, and a complete deletion of the gene. All these mutations were located in various exons of the gene and no relationship was found between the occurrence of PH and a specific position of the mutation. Similarly no relationship has ever been found between the position of the truncating mutation and the features of the NF1 phenotype [53].
There are, therefore, some differences between PH-NF1 and idiopathic and heritable PH. The distinguish features between PH-NF1 and idiopathic and heritable PAH are summarised in table 4.
Pathophysiology of PH in NF1
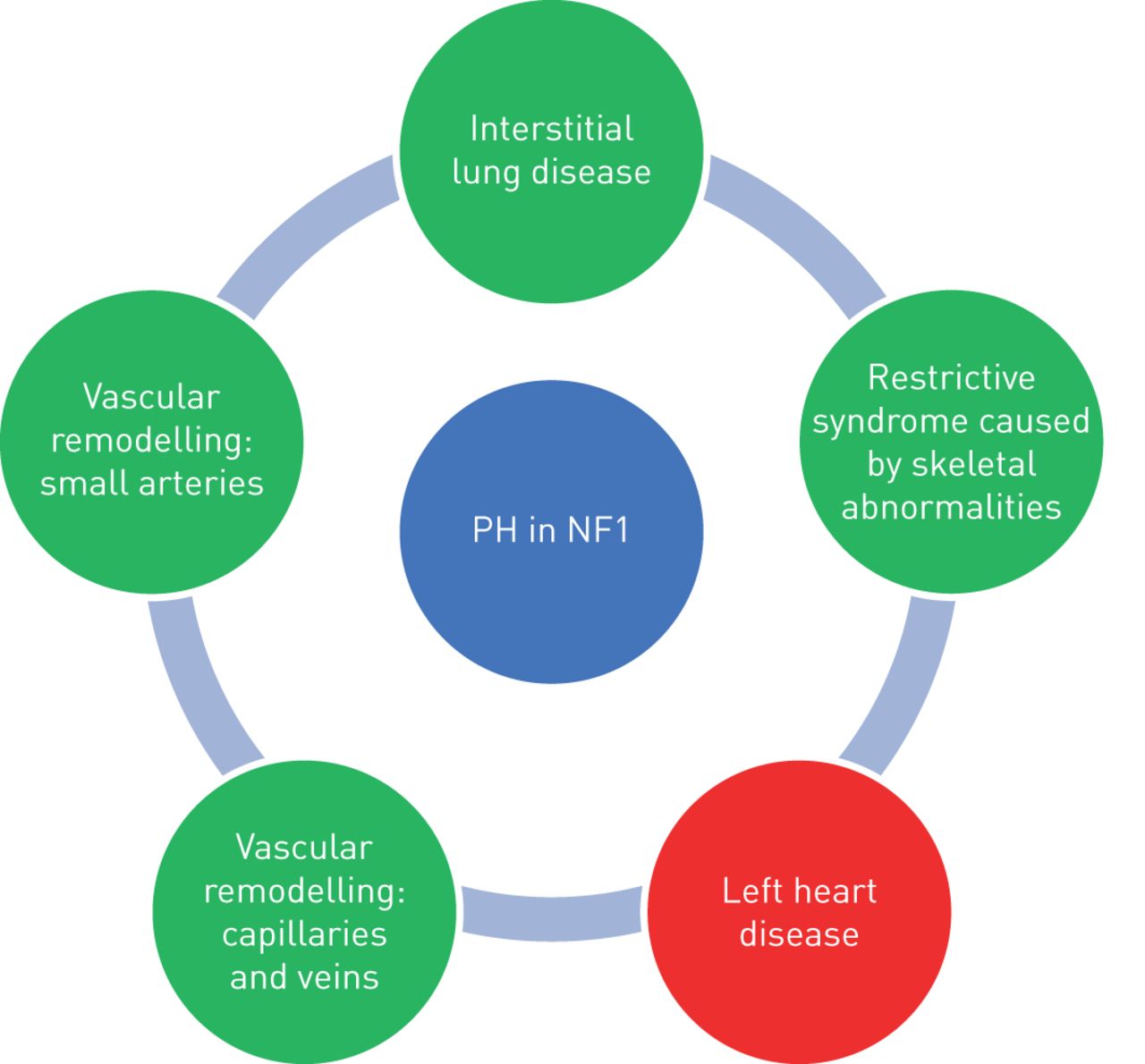
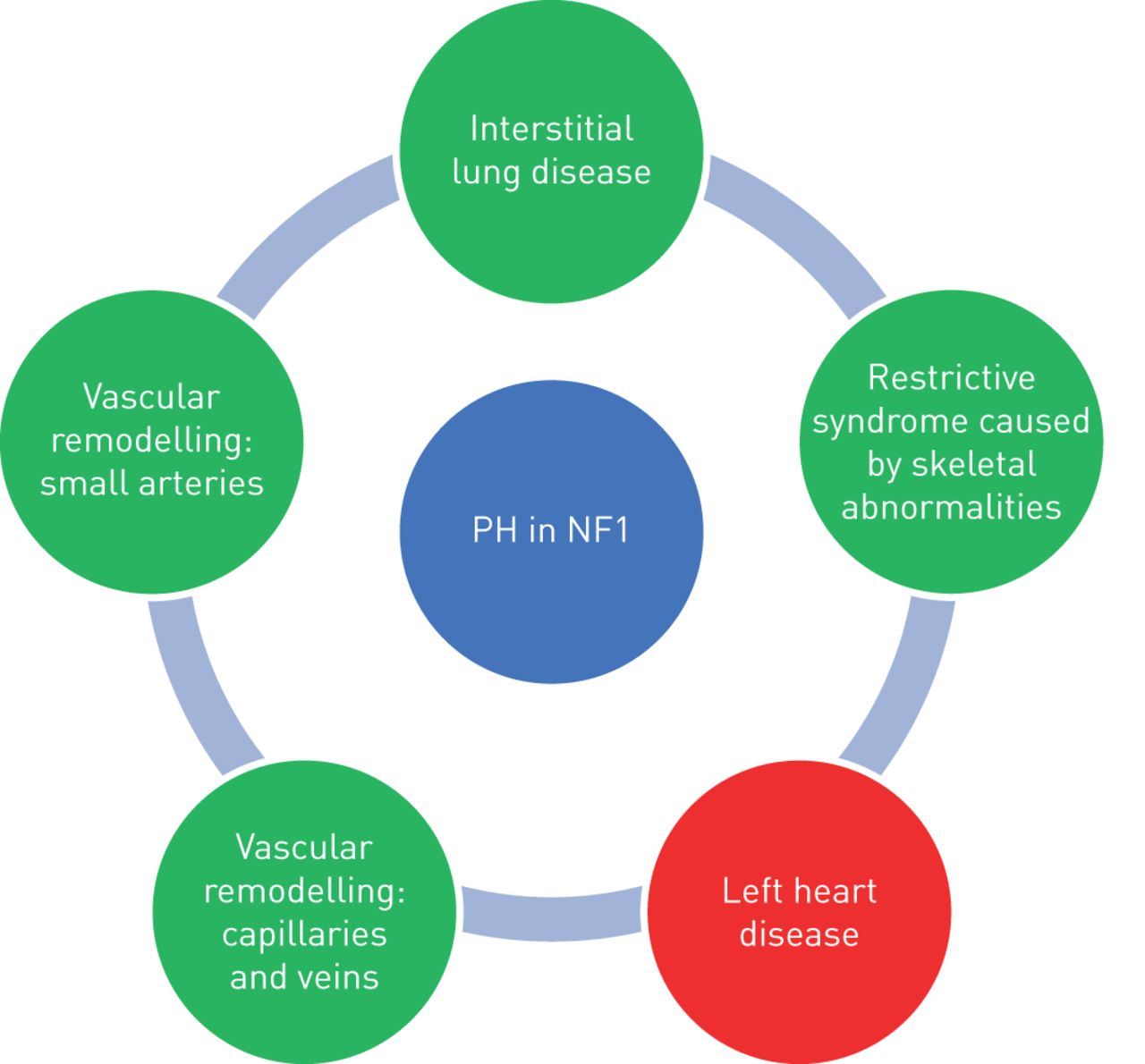
PH-NF1 is classified as group 5 PH, defined as “PH with unclear and/or multifactorial mechanisms”, because the mechanisms of PH remain poorly understood. Indeed, it may include different mechanisms such as lung parenchymal destruction, respiratory insufficiency secondary to restriction caused by skeletal abnormalities, left heart disease, but also pulmonary vascular remodelling of the pulmonary arteries and veins. The different mechanisms of development of PH-NF1 are shown in figure 2.
{kind=link}
{kind=link}
Mechanisms leading to development of pulmonary hypertension (PH) in neurofibromatosis type 1 (NF1). Green indicates pre-capillary hypertension and red indicates post-capillary hypertension.
It is well known that the course of NF1 is often complicated by systemic vasculopathy affecting all the systemic arteries of the body and resulting, for instance, in renovascular hypertension, myocardial infarction, cerebral infarction and ischaemic bowel disease [7]. Lie et al. [7] described three basic types of vascular lesions: 1) zonal intimal vascular smooth muscle cell proliferation in large elastic arteries; 2) intimal vascular smooth muscle cell proliferation, with associated fibrosis and neoangiogenesis of medium-sized elastic and muscular arteries; and 3) plexiform or angiomatoid intimal proliferation in small arteries and arterioles. Knowledge of these systemic vascular complications, has led to the belief that a pulmonary vasculopathy might cause PH-NF1. The description of lung histology which shows the presence of arterial intimal and medial thickening and plexiform lesions are in agreement with this hypothesis, such as the mosaic pattern of lung attenuation seen on the chest HRCT scan of most patients. Veno-occlusive disease has also been suspected or documented [32, 35].
Neurofibromin is known to have a role in tumour suppression. Since neurofibromin is expressed in endothelial and smooth muscle cells of blood vessels [54], its loss is likely to be the key to the development of the vasculopathy associated with NF1. It is interesting that other multiple congenital anomaly syndromes that are due to Ras/MAPK activation by germline mutations (Noonan syndrome, Costello syndrome and Leopard syndrome), are also often associated with PH [55–57]. The mechanisms leading to PH in NF1 and in these diseases may be the same. Molecular mechanisms and cell abnormalities have been studied on systemic endothelial cells showing that loss of NF1 is responsible for an increased proliferation and migration of systemic endothelial cells depending on ERK activation [58]. It has also been shown that systemic vascular smooth muscle cells have an abnormal proliferative phenotype in case of loss of NF1, depending on ERK pathway [59]. In addition, another study showed that there is an increase in reactive oxygen species production in NF1 that could participate in the development of a systemic vasculopathy [60]. However, these mechanisms have not been studied on pulmonary vascular cells.
Moreover, as only a minority of NF1 patients will develop PH, one can hypothesise that a second genetic hit might be necessary to lead to the development of pulmonary vasculopathy. Stewart et al. [26] hypothesised that the pathogenesis of PH-NF1 stems from the loss of heterozygosis of NF1 in pulmonary endothelial cells, the subsequent dysregulation of the RAS pathway, monoclonal expansion of the endothelial cells, abnormal vascular cell proliferation and misguided angiogenesis. Additional unidentified factors may be also involved, such as inflammatory, infectious or autoimmune hits.
Treatment of PH associated with NF1
There is neither long-term data nor recommendations regarding the use of PAH-approved drugs in group 5 PH or in NF1 in particular [1, 61]. Indeed there is only minimal data on the use of these drugs in group 5 PH patients in general and there is the potential for pulmonary venous involvement in this group, which could be aggravated by pulmonary vasodilators [61].
When the diagnosis of PH-NF1 is suspected on transthoracic echocardiography, right heart catheterisation is required to confirm the diagnosis and assess the mechanism (pre- or post-capillary) and severity of the PH. In case of mild PH with severe lung disease, symptomatic treatment should be proposed including long-term oxygen therapy, diuretics and noninvasive ventilation in case of alveolar hypoventilation. In the presence of severe PH without extensive lung involvement, patients should be referred to a PH expert centre and specific PAH treatment should be discussed, based on an understanding of the potential underlying mechanisms and a proper assessment of haemodynamics, with attentive follow-up. Indeed, the benefit/risk ratio of these treatments in PH-NF1 is largely unknown, in particular because of the risk of pulmonary oedema associated with venous and capillary involvement. As PH-NF1 is generally severe, people should be referred early to a lung transplantation centre, with careful assessment of the increased post-transplant cancer risk.
From the known effects of neurofibromin on growth regulation signalling, several currently available agents have been suggested as potentially beneficial in PH-NF1. However, no treatments have been evaluated for this particular indication. Drugs that can inhibit the dysregulation induced by neurofibromin deficiency are potential targets, such as rapamycin, an mTOR inhibitor that has been shown to attenuate PH and neo-intimal formation in rats [62]. A recent study also showed that a stronger inhibition of mTOR by a dual mTORC1/2 inhibitor induced an anti-proliferative effect in NF1-associated plexiform neurofibroma and malignant peripheral nerve sheath tumour cells compared to rapamycin which is only an inhibitor of mTORC1 [63]. Attenuation of the vascular wall proliferation that characterises NF1 using statins (3-hydroxy-3-methylglutarylcoenzyme A reductase inhibitors) is another possible approach, since this class of agent is known to inhibit Ras activity through prevention of its lipid modification [64]. Targeting the MAPK pathway is also promising. Sorafenib is an oral inhibitor of multiple kinases, including Raf, the downstream target of Ras in the MAPK cascade and as described earlier, it improved a refractory PH-NF1 in one case study [31]. However, it has not yet been evaluated in a clinical trial. MEK inhibitors could also be a future strategy, as it has recently been shown that MEK inhibitors, like selumetinib, could be beneficial for plexiform neurofibromas [65] and malignant peripheral nerve sheath tumours in vitro [66]. Clinical trials are warranted to get new effective treatment for PH-NF1 by acting on its molecular mechanisms.
Interstitial lung disease and thoracic complications
Interstitial lung disease
Interstitial lung diseases were the first described lung complications of NF1 [67, 68]. A radiographic study published in 1977 reported that seven out of 70 patients with NF1 had radiographic evidence of “fibrosing alveolitis” with increased interstitial markings, bullous areas of lung destruction or both, and histologic specimens obtained in two patients confirmed lung fibrosis [69]. Bullous and cystic changes, mostly in the upper lobes, were then confirmed to be the most frequent lung manifestations characteristic of NF1, associated or not with interstitial abnormalities [70–72]. In a retrospective single-centre study of 156 NF1 patients, chest radiographs revealed abnormal findings in 70 (44.9%) patients but bilateral interstitial lung infiltrates or cystic airspaces were only demonstrated in eight (5.1%) patients [73]. However, HRCT scanning was not performed in all patients. Another retrospective study, reported three cases of interstitial lung disease associated with NF1 among 55 patients and reviewed 61 other cases described in the literature [44]. Overall, eight (37%) patients had HRCTs demonstrating ground-glass opacities, bibasilar reticular opacities (50%), bullae (50%), cysts (25%) and emphysema (25%); none had honeycombing. In these patients with diffuse lung disease, PFTs showed an obstructive pattern in 43%, restrictive pattern in 37%, and a mixed pattern in 17%, with a decreased diffusion capacity of the lung for carbon monoxide in the vast majority of cases. A group of 14 patients had surgical biopsy results that showed findings of interstitial fibrosis (100%) and interstitial inflammation (93%), consistent with nonspecific interstitial pneumonia. In conclusion, interstitial lung disease is a real complication of NF1 and is characterised by lung cysts or bullae in the upper lobes, diffuse ground-glass opacities sometimes with mosaic pattern and reticular opacities, sometimes with pathologic evidence of fibrosis but without honeycombing on HRCT scans. Another interesting point is that interstitial lung disease has been found to develop only in adulthood whereas NF1 exists from birth.
Other thoracic complications
Thoracic complications of NF1 are multiple and also include the development of airway plexiform neurofibromas, plexiform neurofibromas in the posterior mediastinum, spinal dumb-bell neurofibromas, intercostal neurofibromas, intrathoracic meningoceles, increased risk of pneumothorax or haemothorax [74, 75] and skeletal abnormalities [76]. An increased risk of cancer is known in NF1 and cases of lung cancer have also been described in NF1 although they seem to be rare [77, 78].
Conclusion
PH is a rare but potentially severe complication of NF1. PH-NF1 is classified in group 5 PH, defined as “PH with unclear and/or multifactorial mechanisms”. Severe PH-NF1 is generally associated with lung lesions, mostly cysts or bullae in the upper lobes, diffuse ground-glass opacities sometimes with mosaic pattern and reticular opacities. In one-third of PH-NF1 cases there is no lung parenchymal disease, suggesting specific vascular lesions. There are no guidelines regarding the treatment of PH in these patients and they need to be referred early to expert PH centres and considered for lung transplantation. There are several promising treatments targeting the RAS signalling downstream pathways which will require clinical trials.
Footnotes
Number 1 in the Series “Group 5 Pulmonary Hypertension” Edited by Yochai Adir and Laurent Savale
Provenance: Commissioned article, peer reviewed.
Conflict of interest: E-M. Jutant has nothing to disclose.
Conflict of interest: B. Girerd has nothing to disclose.
Conflict of interest: X. Jäis reports grants, personal fees and non-financial support from Actelion, GSK, Bayer and MSD, outside the submitted work.
Conflict of interest: L. Savale reports grants and personal fees from Actelion, grants, personal fees and non-financial support from MSD and Bayer, and personal fees and non-financial support from GSK, outside the submitted work.
Conflict of interest: C. O'Connell has nothing to disclose.
Conflict of interest: F. Perros has nothing to disclose.
Conflict of interest: O. Sitbon reports grants, personal fees and non-financial support from Actelion Pharmaceuticals, Merck and GSK, grants and personal fees from Bayer, and personal fees from Arena Pharmaceuticals and Acceleron Pharmaceuticals, outside the submitted work.
Conflict of interest: M. Humbert reports personal fees from Actelion, Merck and United Therapeutics, and grants and personal fees from Bayer and GSK, outside the submitted work.
Conflict of interest: D. Montani reports grants and personal fees from Actelion and personal fees from BMS, GSK, MSD and Pfizer, outside the submitted work.
- Received June 2, 2018.
- Accepted July 28, 2018.
- Copyright ©ERS 2018.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










