





Abstract
Bronchiectasis is a chronic respiratory disease characterised by a syndrome of productive cough and recurrent respiratory infections due to permanent dilatation of the bronchi. Bronchiectasis represents the final common pathway of different disorders, some of which may require specific treatment. Therefore, promptly identifying the aetiology of bronchiectasis is recommended by the European Respiratory Society guidelines. The clinical history and high-resolution computed tomography (HRCT) features can be useful to detect the underlying causes. Despite a strong focus on this aspect of treatment a high proportion of patients remain classified as “idiopathic”. Important underlying conditions that are treatable are frequently not identified for prolonged periods of time.
The European Respiratory Society guidelines for bronchiectasis recommend a minimal bundle of tests for diagnosing the cause of bronchiectasis, consisting of immunoglobulins, testing for allergic bronchopulmonary aspergillosis and full blood count. Other testing is recommended to be conducted based on the clinical history, radiological features and severity of disease. Therefore it is essential to teach clinicians how to recognise the “clinical phenotypes” of bronchiectasis that require specific testing.
This article will present the initial investigation and management of bronchiectasis focussing particularly on the HRCT features and clinical features that allow recognition of specific causes.
Abstract
Bronchiectasis is a heterogeneous disease with diverse clinical presentation. Careful history, review of radiological features and laboratory testing are required to identify the underlying diagnosis. http://ow.ly/RDF730koTxu
Introduction
Bronchiectasis is a progressive respiratory disease characterised by permanent dilatation of the bronchi and associated with a clinical syndrome of cough, sputum production and recurrent respiratory infections [1]. The causes of bronchiectasis are varied with important differences between the presentation and natural history of the disease depending on aetiology.
Bronchiectasis is increasing in prevalence with current rates estimated between 53 and 566 cases per 100 000 inhabitants depending on the population studied [2, 3]. These differences in reported prevalence may be due to the long period of neglect and growing awareness or could represent a true rise in prevalence. It should therefore be expected that cases of bronchiectasis will be encountered more frequently by the general physician, as well as the respiratory specialist. Bronchiectasis is a heterogenous disease with many causes and associations. The most commonly associated conditions are shown in table 1. Although the final clinical syndrome is similar, there are many clinical and radiological features which give clues as to aetiology. The presentation of post-infective bronchiectasis can be very different to the presentation of chronic obstructive pulmonary disease (COPD)-related bronchiectasis and the features of a computed tomography (CT) scan of post-tuberculous bronchiectasis are different to the features seen with nontuberculous mycobacteria (NTM) related disease, for example. Identifying the underlying cause accurately and quickly is a key recommendation of international guidelines, as many causes of bronchiectasis are treatable or have specific prognostic implications (table 1).
Aetiologies of bronchiectasis
Our understanding of the pathophysiology of bronchiectasis is limited. The so-called “vicious cycle hypothesis” first proposed in 1986 by Cole [4] remains central to our understanding. The key components of the disease are chronic inflammation, impaired mucociliary clearance, chronic bronchial infection and structural lung damage. Chronic airways infection, most frequently with Haemophilus influenzae and Pseudomonas aeruginosa, stimulates and sustains lung neutrophilic inflammation and is related with a higher frequency of exacerbations, worse quality of life and increased mortality [5]. This is particularly the case with P. aeruginosa infection where chronic infection is associated with a three-fold increase in mortality and seven-fold increase in hospitalisation [6].
Recognised aetiologies include post-infection, COPD, primary ciliary dyskinesia (PCD), allergic bronchopulmonary aspergillosis (ABPA), NTM infections, immune deficiencies and connective tissue diseases [7]. However, despite extensive testing, up to 53% of patients may have no identifiable cause and the diagnosis of idiopathic bronchiectasis remains common [8].
The recent European Respiratory Society (ERS) guidelines suggest the following minimum bundle of aetiological tests to perform in adults with a new diagnosis of bronchiectasis: measurement of differential blood count, immunoglobulins (IgA, IgM and IgG) and screening for ABPA (total IgE, specific IgE to Aspergillus, IgG to Aspergillus and eosinophil count). Additional tests may be appropriate in specific clinical features or in patients with severe or rapidly progressive disease. Sputum culture is recommended for monitoring bacterial infections and when NTM infection is suspected [9]. Standardised tests are important to seek causes of underlying bronchiectasis because they lead to a change in treatment in 7–37% of cases [7, 8, 10, 11].
Chest high-resolution computed tomography (HRCT) features can be useful to detect the underlying causes. HRCT is now the accepted standard to establish the diagnosis of bronchiectasis [1]. The prerequisite is the identification of dilation of the airways, seen as an increased ratio between the internal lumen of a bronchus and its immediately adjacent pulmonary artery. The lack of normal tapering, mucus plugging, nodules, bronchial wall thickening, “tree-in-bud” pattern, lung volume loss and mosaicism pattern are all additional features useful to support a diagnosis of bronchiectasis. Furthermore, all these signs can be associated with particular distributions of bronchiectasis and can guide us to a specific cause [12].
As the ERS guidelines recommend only a small number of tests are performed routinely, it is important that clinicians know how to recognise other treatable causes and “phenotypes”, as listed in table 1. This article presents examples of clinical phenotypes, integrating history and HRCT features to illustrate the importance of identifying the underlying cause of bronchiectasis and key components of management.
Case 1
Case 1 is a 42-year-old man. He works as a gardener but has been finding it difficult to maintain his business recently due to recurrent respiratory infections. He has had a long history of respiratory problems starting in early childhood. He thinks he was told that he had asthma and previously used an inhaler but stopped due to it being ineffective. He struggled at school due to frequent absence due to “chest infections”. He is unaware of any neonatal issues but believes that he was born at home without complications and is unsure of any previous tests he has had as he is now estranged from his parents. He believes he has a cousin with a “lung disease”.
He tends to cough most days and has three to four significant chest infections per year. He has often struggled to gain weight. He is married but has not had any children.
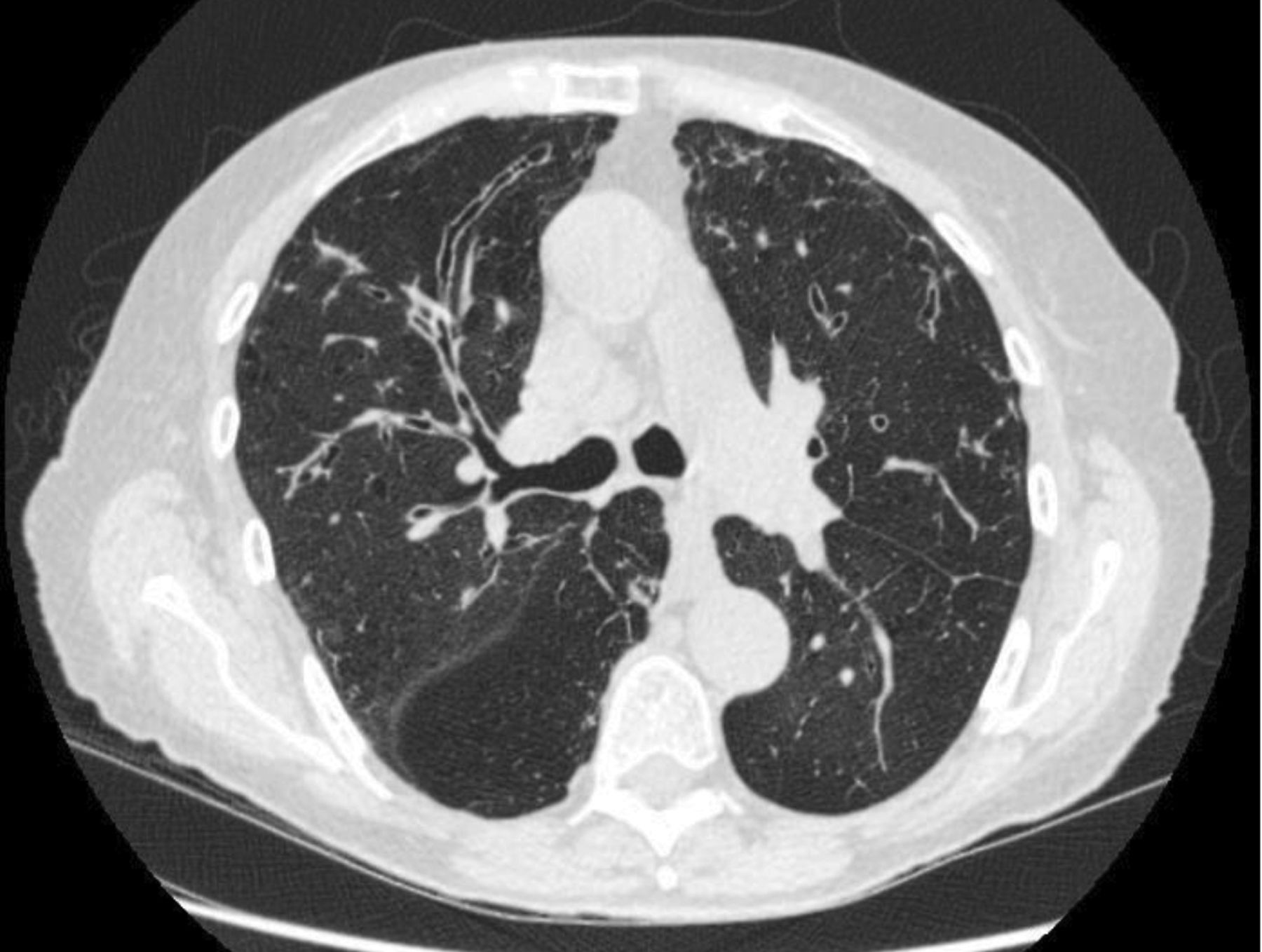
His initial screening blood tests did not reveal any abnormalities. His CT scan showed extensive cystic and varicose bronchiectasis affecting all lobes including the upper lobes (figure 1). His sputum culture taken in clinical stability grew P. aeruginosa. No previous cultures are available to determine the duration of this infection. Due to his family history, infertility, severity of disease and young age of onset cystic fibrosis (CF) genetics were performed. His genotype was F508del/R117H, with a sweat chloride of 73 mEq·L−1. A diagnosis of CF was made.

Varicose and cystic bronchiectasis with mucus plugging in upper lobes.
Cystic fibrosis
CF is the most common life-threatening autosomal recessive disease in the USA and Europe [13, 14].
CF is a multisystem disorder caused by mutations in the gene that encodes the CF transmembrane conductance regulator (CFTR) protein, a chloride channel expressed in epithelial cells [15]. More than 2000 CFTR mutations have been identified to date, but only the functional importance of a small number is known to cause the disease [16].
Clinical manifestations of CF can be very different between patients according to their genotype and the consequent highly variable levels of CFTR dysfunction [15, 17].
Bronchiectasis associated with cough, sputum production and recurrent respiratory infections are the hallmark of CF lung disease. An upper lobe predominant distribution of cylindrical, cystic and varicose bronchiectasis associated with airway wall thickening, mucus plugging and parenchymal opacities on a HRCT scan should raise the suspicion of CF disease [18]. The presence of nasal polyposis and/or chronic rhinosinusitis, recurrent pancreatitis, malabsorption, diabetes, osteoporosis and male infertility are other typical features of CF and the diagnosis of CF should be considered in any adult who presents with these signs and symptoms, especially if they started in childhood.
According to the recent guidelines published by the Cystic Fibrosis Foundation in the USA [19], CF is diagnosed when an individual has both a clinical presentation of the disease and evidence of biochemical and genetic markers of CFTR dysfunction. If an individual with clinical features of the disease has a concentration of chloride >60 mmol·L−1 at the sweat test or a concentration in the intermediate range (30–59 mmol·L−1) but two disease-causing CFTR mutations the diagnosis of CF can be made. If the CFTR genotype is undefined, CFTR physiologic tests, such as nasal potential difference and intestinal current measurement, should be performed.
However, because of widespread CF newborn screening based on the measurement of immunoreactive trypsinogen in blood spots, 55.5–73% of new CF diagnoses in Europe now occur in asymptomatic or minimally symptomatic infants [20–22], so that it has become unusual to diagnose CF in patients with classic symptoms of respiratory disease and emaciation at an advanced age [23]. Nevertheless, the diagnosis can be challenging or inconclusive in some individuals [24]. For positive screened individuals with inconclusive CFTR functional and genetic testing the CFTR-related metabolic syndrome definition should be used, whereas for non-screened individuals presenting with monosymptomatic clinical entity associated with CFTR dysfunction that does not fulfil the diagnostic criteria for CF, a diagnosis of CFTR-related disorder should be considered [19].
Diagnosing CF is an important goal in adults with bronchiectasis. Around 7% of patients with CF are diagnosed as adults [23]. Patients affected by CF should be referred to a CF specialist centre because CF has a distinct pathophysiology, prognosis and treatment pathway. In many countries, CF patients have more access to respiratory therapies than patients with non-CF bronchiectasis. Furthermore, new CFTR modulator therapies can be provided to patients with specific mutations in order to improve their clinical outcomes.
The presence of a single CFTR mutation in the presence of a normal sweat test is relatively common in bronchiectasis and its clinical significance is uncertain.
The authors' current practice is to screen for CF in all patients presenting with bronchiectasis before the age of 50 years, as in this case, and all patients with bronchiectasis symptoms onset during childhood irrespective of the age of presentation. In addition, the presence of upper lobe disease, Staphylococcus aureus or P. aeruginosa in sputum, or extrapulmonary features such as malabsorption, pancreatitis or infertility should also prompt CF testing irrespective of the age of the patient.
Case 2
Case 2 is a 45-year-old architect. He has had asthma since childhood. It has generally been well controlled throughout most of his adult life with only one or two exacerbations and no hospital admissions. In recent years he has noticed a decline in his exercise tolerance and an increase in cough which has become productive of purulent sputum with occasional thick/solid components. He has had several significant respiratory exacerbations which have not responded well to standard steroid and antibiotic treatment. One of these episodes was so severe he required emergency hospital admission due to respiratory distress. This improved when he managed to expectorate some very thick sputum. He was noted to have variable pulmonary infiltrates on chest radiographs during these episodes.
He attended a general respiratory clinic. A CT scan showed significant central bronchiectasis (figure 2). He was noted to have a marked eosinophilia on initial blood tests. This prompted Aspergillus serological testing. His total IgE was >2000 IU·mL−1, his Aspergillus IgE was 750 IU·mL−1 and Aspergillus IgG was 45 IU·mL−1. A diagnosis of ABPA was made and he was commenced on high-dose oral corticosteroids.
Central tubular bronchiectasis in upper lobes.
Allergic bronchopulmonary aspergillosis
ABPA is an inflammatory disease caused by hypersensitivity to the ubiquitous fungus Aspergillus fumigatus. [25].
ABPA occurs most commonly in patients with asthma [26] and CF [27], but many patients do not have a history of asthma. ABPA is the cause of 1–10% of cases of bronchiectasis [8, 28, 29], but it can also complicate pre-existing bronchiectasis. Most ABPA cases occur in the third and fourth decade without a sex predilection.
Clinically, patients affected by ABPA may present with symptoms such as malaise, weight loss, low-grade fever, cough, purulent sputum containing brownish-black mucus plugs, pleuritic chest pains and haemoptysis [30].
There is not a single test to diagnose ABPA or a universally recognised set of criteria. Integration of patient's history, clinical, radiological and serological features is used to diagnose ABPA. When ABPA is suspected, total IgE, specific IgE to Aspergillus or Aspergillus skin prick testing, IgG to Aspergillus and eosinophil count should be performed [9]. Total serum IgE levels >1000 IU·mL−1 are the hallmark of ABPA and total IgE levels are also the most useful test for follow-up [30]. Caution is recommended in interpreting total IgE levels in patients recently treated with oral corticosteroids as “partially treated” ABPA may result in lower levels. A. fumigatus-specific IgE levels are also elevated and more sensitive than skin-prick test to Aspergillus antigen [31]. Peripheral eosinophilia and raised IgG antibodies against Aspergillus are also supportive of diagnosis [30].
HRCT is particularly useful in identifying ABPA as central bronchiectasis is a classical finding, even though peripheral bronchiectasis may occur [32]. The upper lobes are most frequently affected [33] but bronchiectasis may be present in all lobes [34]. Additional findings on HRCT are mucus plugging with “finger-in-glove” appearance, transient consolidation, centrilobular nodules associated with tree-in-bud, atelectasis, mosaicism due to air trapping on expiration and fibrosis in end-stage disease [30]. In some cases though, ABPA without bronchiectasis can be recognised [35]. Distinguishing between ABPA diagnosed serologically (ABPA-S) and ABPA with central bronchiectasis (ABPA-CB) may have prognostic implications: ABPA-S may be a more benign phenotype of ABPA, but most experts consider it a precursor of ABPA-CB [36].
The clinical course of ABPA is variable. There are five recognised stages of ABPA: stage I defines new, active ABPA; stage II is clinical and serological remission; stage III is recurrent active ABPA; stage IV defines patients with chronic, steroid-dependent ABPA; and stage V is end-stage disease with fibrocavitary lesions [37]. ABPA is important to identify as progressive lung damage occurs rarely once treatment is started [36].
Many drugs have been tried in the treatment of ABPA, such as: systemic and inhaled corticosteroids with the aim to reduce the inflammatory response; antifungal agents, that decrease the antigen burden and subsequent immune response; and omalizumab, a monoclonal antibody directed against IgE [38]. The initial treatment for ABPA is usually with oral corticosteroids [1]. Treatment is typically started at high doses, e.g. 40 or 50 mg daily, with weaning of the dose over several months with monitoring of total IgE and clinical symptoms.
ABPA remission is defined by improvement in symptoms, decrease in total serum IgE level, resolution of radiographic infiltrates and improvement in lung function [39]. Failure to respond to therapy should prompt consideration of comorbidities or alternative diagnoses. Infective exacerbations of bronchiectasis are associated with cough, wheeze and mucus plugging so that it may be challenging to discern if the primary driver of sputum is bacterial or allergic.
The role of antifungal treatment in ABPA is controversial. A double-blind randomised controlled trial previously demonstrated that treatment response, defined as a reduction of corticosteroid dose of >50%, was more rapid in patients taking a combination of steroids plus itraconazole compared to corticosteroids alone. This study suggests that itraconazole can be used as a steroid sparing agent [40]. Many clinicians use itraconazole in combination with corticosteroids as initial therapy for ABPA. Evidence for this approach is limited. A recent randomised controlled trial with 131 patients included found a similar rate of response in terms of IgE and exacerbations with initial therapy using prednisolone alone versus itraconazole alone, which does suggest that reducing fungal burden may be important alongside reducing inflammation [41]. We do not currently advocate treatment with antifungals alone. Concerns with antifungals include liver toxicity, photosensitivity and drug–drug interactions. Patients taking itraconazole should not take proton-pump inhibitors since they require an acidic environment to be absorbed.
Case 3
Case 3 is a 19-year-old student. He arrived in the UK from Pakistan aged 13 years. He has had respiratory problems since early childhood. His grandparents describe him as a small child with chronic cough since birth, as well as recurrent ear and sinus infections which have led to partial hearing loss. His brother and one of his cousins are similarly affected. His parents are first cousins. Since arriving in the UK he has had two hospital admissions with pneumonia. He has had a provisional diagnosis of asthma due to frequent symptoms even when well, but standard treatment has not been effective.
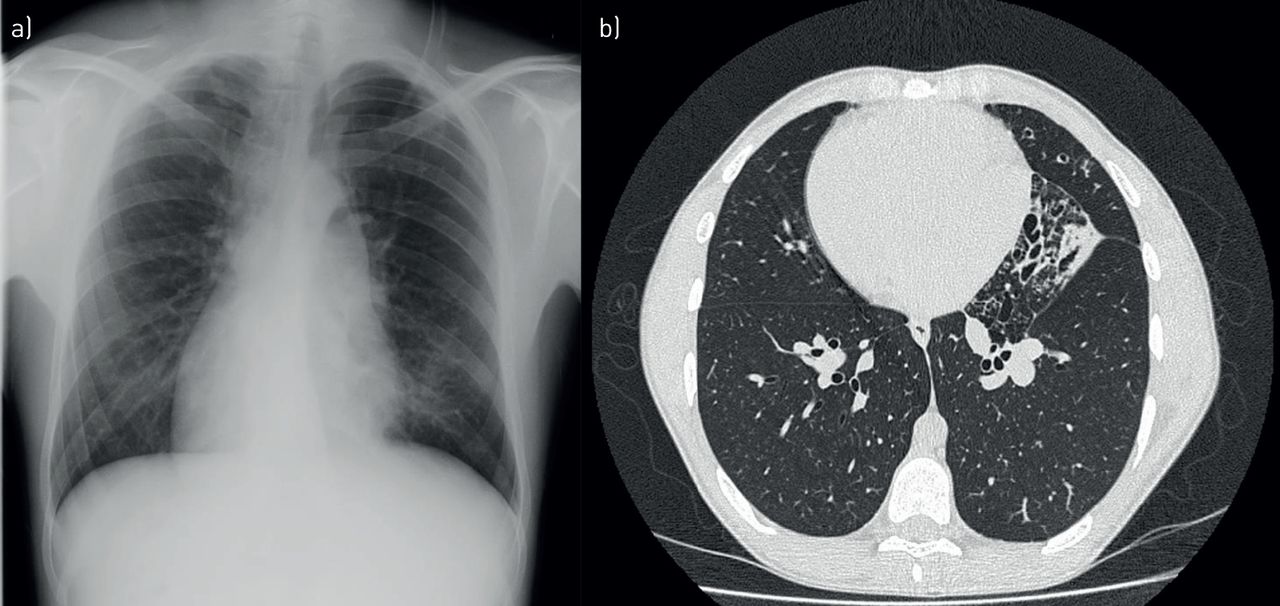
Examination revealed dextrocardia which was confirmed on radiographs and CT. CT scans also showed minor lingula bronchiectasis (figure 3). He grew H. influenzae on several occasions on sputum culture. Due to his age and family history, he was screened for CF (which was negative) and PCD. His nasal nitric oxide (nNO) was inconclusive and he went on to have a high-speed video analysis (HSVA) examination of nasal epithelial cells. This demonstrated static cilia. Electron microscopy and genetic testing revealed an outer dynein arm defect and biallelic pathogenic mutations in DHAH5 which confirmed the diagnosis of PCD. His sibling was also later diagnosed.

a) Chest radiograph showing dextrocardia. b) High-resolution computed tomography showing cylindrical bronchiectasis in the lingula.
Primary ciliary dyskinesia
PCD is a rare genetic disorder with an estimated prevalence of 1:10 000 [42]. PCD is caused by recessive autosomal or X-linked mutations in one of more than 35 genes which lead to functional and/or structural defects of cilia [43]. Motile cilia are specialised structures present in many tissues, so their immotility or ineffective beating results in a systemic disease with heterogeneous features. In upper and lower respiratory airways, failure of motile cilia leads to poor mucociliary clearance, which triggers a vicious cycle of inflammation and infection that leads to chronic rhinosinusitis and otitis media, progressive airway obstruction, bronchiectasis and ultimately respiratory failure [44]. A history of neonatal respiratory distress is often reported [45]. Since embryonic nodal cilia can also be defective, ∼50% of patients have situs inversus totalis or heterotaxy. When situs inversus is accompanied by chronic sinusitis and bronchiectasis, this is known as Kartagener's syndrome [44]. Sperm flagella and cilia of the fallopian tubes share common axonemal structures with motile cilia, so a proportion of PCD-affected males and females are infertile and female patients that are fertile are at a higher risk of complications such as ectopic pregnancy [46].
The PCD diagnostic pathway is complex and some of its tests are very expensive or require a high level of expertise, so only a few tertiary centres can perform all of them. According to the 2017 ERS PCD guidelines, a diagnostic PCD step-wise approach includes firstly the measurement of nNO and the performance of nasal brushing for the evaluation of ciliary beat frequency and pattern with HSVA. The evaluation of ciliary ultrastructural defects with transmission electron microscopy or genetic testing can be used to confirm the diagnosis. Neither modality is able to detect all cases of PCD, with ∼80% and 70% of specificity, respectively. Therefore, in cases where diagnosis remains inconclusive, repeating the high-speed video microscopy on a new sample or after in vitro culture of the airway epithelium is suggested. Detection of presence or absence of ciliary proteins on ciliated respiratory epithelial cells by immunofluorescence is also increasingly used [47]. In the ERS bronchiectasis guidelines, testing for PCD is limited to patients with clinical features consistent with the disease (persistent productive cough since childhood, chronic rhinosinusitis, chronic middle ear disease with or without hearing loss, situs anomalies, congenital cardiac defects and a history of neonatal respiratory distress or neonatal intensive care admittance in term infants) [9]. All these clinical factors are included in the PICADAR score, a simple diagnostic tool to predict whether symptomatic patients have PCD [48]. PICADAR score and nNO are suitable and easy-to-perform tests in respiratory clinics, whereas further tests should be performed in PCD or tertiary centres with high expertise. Importantly, although nNO is often used as a screening test, it can be normal or inconclusive in some cases of PCD, as in this case. Therefore, where there is a high index of suspicion as in this case, further investigations should be performed irrespective of the nNO result.
HRCT features may be helpful in raising the suspicion of PCD. Bronchiectasis in PCD is predominantly in the middle lobe, lingula and lower lobes, with a central or diffuse distribution. As expected from the underlying pathophysiology of the disease, the severity of bronchiectasis increases with age, whereas data regarding the correlation between HRCT features and pulmonary function are conflicting [49, 50]. Besides lung parenchymal alterations, situs inversus, heterotaxy and congenital heart disease could support the suspicion of PCD. Pectus excavatum is identified in 9% of cases [49].
Investigation and diagnosis of PCD is an important goal because it is a multi-system disease which benefits from a specific and multidisciplinary team approach, including genetic and reproductive counselling, in a PCD referral centre [1]. Although airway clearance is an important aspect of the management of all bronchiectasis patients, it is particularly emphasised in the management of patients with PCD. The importance of making the diagnosis is emphasised by data showing worse lung function and higher rates of P. aeruginosa infection the later patients are diagnosed [51–55].
Case 4
Case 4 is a 77-year-old retired librarian. She has had cough for many years, although she has only sought medical attention recently as she has developed more systemic symptoms. Her new symptoms are fatigue, weight loss and fever. A chest CT scan was performed looking for a possible occult malignancy and bronchiectasis was found (figure 4).

Cylindrical bronchiectasis and tree-in-bud pattern in lower lobes and middle lobe.
Following the CT scan features of cylindrical bronchiectasis and tree-in-bud pattern in middle and lower lobes, she attended a respiratory clinic. A sputum sample was sent and a positive acid-alcohol-fast bacilli staining was found. After 4 weeks Mycobacterium avium complex (MAC) grew on the culture. Several subsequent cultures were also positive. As NTM can complicate pre-existing bronchiectasis as well as being a cause of bronchiectasis, further testing for immunodeficiency, ABPA and other causes was performed. No abnormalities were found. In view of the clinical history, radiological findings and absence of an alternative cause, a diagnosis of bronchiectasis due to NTM lung disease was made.
Nontuberculous mycobacteria
NTM are ubiquitous organisms in the environment and can be inhaled or ingested from water, soil and dust with different consequences according to individual and organism characteristics.
The exact prevalence of NTM disease is difficult to obtain because reporting these infections is not mandatory in many countries and discriminating between colonisation and active infection can be challenging. Certainly, the recovery of NTM from the respiratory tract is increasing as a result of a real growing prevalence of pulmonary disease due to these organisms [56], but also because of improving microbiological techniques and the increasing attention to this topic. In a systematic review, the overall prevalence of NTM in patients with bronchiectasis was 9.3%, with higher rates in larger cohorts and in Asian populations [57]. The most frequent species is MAC (which includes M. intracellulare, M. avium and M. chimaera), with a percentage up to 80% of all NTM lung disease cases [58, 59] whereas the isolation of other species, e.g. M. abscessus, M. kansasii, M. chelonae and M. fortuitum, varies from study to study.
The outcome of respiratory exposure to NTM, colonisation or pulmonary disease, probably depends on a complex relationship between host-related factors (genetics, immune status and lung damage), exposure-related factors (duration, number of organisms, inhaled or ingested particle size) and organism-related factors (capability of mycobacteria to cause pulmonary disease, e.g. M. malmoense has more pathogenicity than M. chelonae [60]). NTM can be both a cause and a consequence of bronchiectasis. NTM can cause bronchiectasis by destroying the bronchial wall and bronchiectasis can predispose to NTM colonisation/disease due to an impairment of bronchial anatomy and local host defence [61, 62].
The diagnosis of NTM pulmonary disease is based on the presence of clinical criteria (respiratory symptoms consistent with the diagnosis), radiological features consistent with NTM, exclusion of other diagnoses and microbiological criteria (a positive culture from one bronchial lavage, at least two NTM-positive sputum cultures or a lung biopsy with mycobacterial histopathologic features plus a NTM-positive culture) [63]. NTM pulmonary disease is often challenging to diagnose, due to the presence of organisms in the environment and the consequent possibility of sample contamination and the wide spectrum of clinical manifestations. If a diagnosis of NTM pulmonary disease is difficult in healthy individuals, it is even more difficult in bronchiectasis patients, where distinguishing between bronchiectasis-related and NTM-related symptoms can be challenging. According to the ERS guidelines, three sequential daily sputum cultures for mycobacteria or a single bronchoalveolar lavage should be considered in patients with persistent fever, weight loss, haemoptysis, symptoms non-responsive to standard therapy or rapid clinical deterioration [9]. Up to 50% of patients will not produce sputum spontaneously and such patients should be investigated by bronchoscopy or sputum induction.
HRCT features should also be investigated when NTM pulmonary disease is suspected. Two major radiological patterns are related to NTM infection: the nodular/bronchiectatic and fibrocavitary forms. The first one is characterised by multiple small centrilobular nodules and cylindrical bronchiectasis, especially localised in middle lobe and lingula. [64]. This pattern is frequently associated with MAC infection [65] and with the “Lady Windermere syndrome”: women with elderly age, low body mass index and chronic cough [66]. The fibrocavitary form is characterised by increased opacity areas and cavitations, usually in the upper lobes, with or without calcifications. In addition, apical pleural thickening and fibrosis with volume loss can be found. Pleural effusion, adenopathies and lower lobes involvement are uncommon [67]. This pattern is frequently associated with M. abscessus, M. chelonae and M. kansasii infections [65]. The underlying pulmonary disease in these patients is often advanced COPD.
Once a diagnosis of NTM disease is established, the institution of therapy is not always mandatory. Since NTM treatment is usually prolonged and based on multiple drugs with significant side effects, a careful evaluation of risks and benefits should be made. Considering NTM species, patient's conditions, radiological pattern and disease severity is crucial in the decision-making process. In the case of NTM treatment, patients should be closely monitored with visits, sputum cultures, lung function and blood tests to assess the response to therapy and possible side effects. Follow-up should also be prolonged after treatment since relapse or new infections are possible and the underlying bronchiectasis requires long-term management. In case of no treatment, patients should be monitored to evaluate disease progression.
This patient was treated with rifampicin 600 mg daily, ethambutol 15 mg·kg−1 daily and clarithromycin 500 mg twice daily. This, sometimes with the addition of intravenous amikacin for 3 months or nebulised amikacin, is the regimen recommended for severe MAC pulmonary disease where acid-fast bacilli is smear positive or there are severe symptoms of systemic illness or cavitation. Intermittent therapy three times per week (rifampicin 600 mg three times per week, ethambutol 25 mg·kg−1 three times per week and azithromycin or clarithromycin three times per week) are recommended in patients with mild disease who are smear negative with the absence of cavitation and no systemic symptoms. Patients who have intolerance to drugs or refractory disease should be referred to an NTM speciality clinic.
Case 5
A 66-year-old woman with established idiopathic bronchiectasis has had three to four exacerbations per year for the past 3 years despite performing daily chest physiotherapy. Testing for NTM, ABPA and other complications were negative, but sputum shows persistent infection with P. aeruginosa. She produces large volumes of sputum daily despite performing the active cycle of breathing technique.
This is one of the most common presentations of bronchiectasis, as exacerbations are one of the most important manifestations of bronchiectasis and P. aeruginosa is the most frequent organism in severe bronchiectasis worldwide. This patient could present any of the CT scans shown above (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
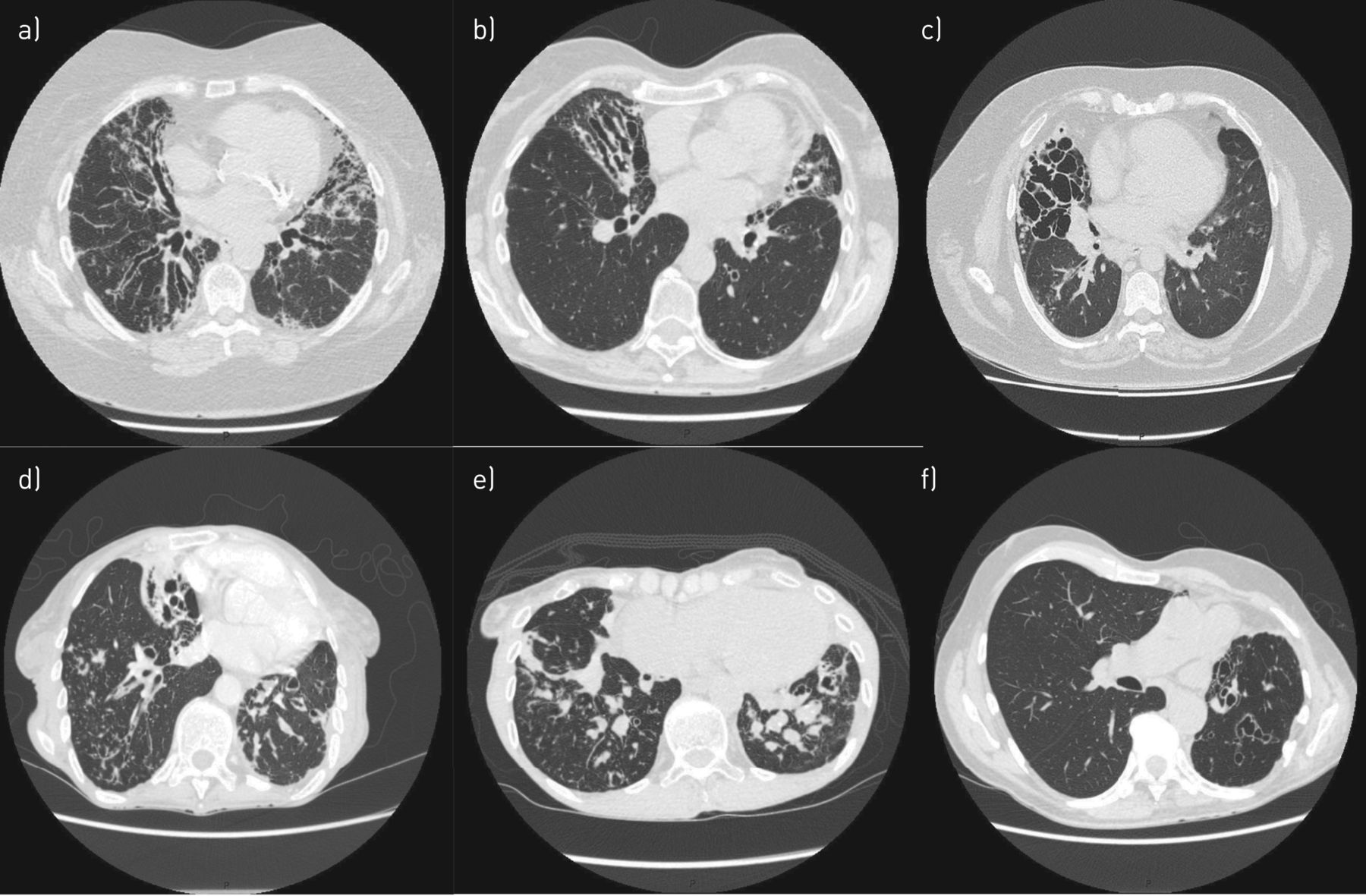
Different radiological “phenotypes” of bronchiectasis. a) Tubular bronchiectasis in lower lobes, b) varicose bronchiectasis in lower lobes, c) cystic bronchiectasis in right lower lobe, d) cystic bronchiectasis with tree-in-bud pattern and mucus plugging in lower lobes (cavitation in right lower lobe), e) mucus plugging and tree-in-bud pattern in lower lobes, f) varicose bronchiectasis in upper lobes and lingula.
Cylindrical bronchiectasis is the most common morphological pattern identified on CT scans [68] and is recognised by an abnormal dilatation of bronchus with uniform calibre and lack of tapering at the periphery, producing a tramline (tram track sign) (figure 5a). Varicose bronchiectasis has no regular form or size, with distortion and irregular bulging (figure 5b), whereas cystic bronchiectasis is a saccular dilatation with ballooned cutline that can be traced almost to the pleura (figure 5c). In the context of bronchiectasis cavitation can also be seen, especially in NTM pulmonary disease or previous M. tuberculosis infection (figure 5d). Another frequent finding is mucus plugging, the presence of mucoid secretions in peripheral airways (figure 5e), that can produce V- and Y-opacities, the so-called tree-in-bud pattern (figures 5d and e). The distribution and extension of bronchiectasis are also key components in bronchiectasis evaluation. For instance, localised bronchiectasis, especially in a single lobe, should raise the suspicion of obstructive or post-infective bronchiectasis (figure 5f).
Management of frequent exacerbations
Frequent exacerbations should prompt a review of all aspects of management including reviewing current airway clearance regime, repeat sputum microbiology and repeat testing for NTM, ABPA and ensuring the all possible treatable causes and comorbidities have been identified.
Intensification of treatment should address all aspects of the vicious cycle including targeting airway infection, airway clearance and airway structural damage. Interventions that have been shown to reduce exacerbations and improve quality of life in either controlled trials or observational studies include macrolides [69–71], inhaled antibiotics [72–74], mucoactive drugs (e.g. hypertonic saline) [75, 76], airway clearance [77, 78] and pulmonary rehabilitation [79, 80]. All of these interventions were recommended in appropriate patients in the 2017 ERS guidelines [9].
The first-line recommendation for P. aeruginosa with frequent exacerbations is an inhaled antibiotic [9] and this patient was commenced on twice daily colistin treatment. Airway clearance was optimised through an appointment with a respiratory physiotherapy and nebulised saline was added to her daily regimen. She was referred to and completed pulmonary rehabilitation. These interventions resulted in a reduction of her exacerbation frequency and an improvement in quality of life. Important issues with inhaled antibiotics include an approximately 10% risk of bronchospasm [72] and the burden of administering a daily nebulised therapy that takes some time. Antibiotic resistance is also a theoretical risk with any long-term antimicrobial treatment. Treatment of these patients with multiple nebulised therapies plus airway clearance results in a large burden of treatment and increases the risk of non-adherence. Macrolides are a reasonable alternative with a lower treatment burden but have less evidence in the context of chronic P. aeruginosa infection [69–71]. Furthermore, it is important to exclude NTM prior to commencing macrolide therapy to avoid inducing resistance. A detailed discussion of management of bronchiectasis is beyond the scope of this review. Further information on treatment including antibiotic therapy, thoracic surgery, physiotherapy management and treatment of acute exacerbations can be found in the 2017 ERS guidelines [9].
Conclusion
Bronchiectasis is no longer a neglected disease. Interest and awareness are increasing in scientific and patient communities. Chest physiotherapy and antibiotic treatment when an exacerbation occurs remain the cornerstone of bronchiectasis therapy. Discovering the underlying aetiology of bronchiectasis could make a real difference in the management and prognosis of patients and could delay the progression of lung involvement when treated. Since routine testing of all patients for all possible underlying disorders has not been shown to be cost-effective, current guidelines recommend using clinical judgement and recognition of patient phenotypes to guide testing for disorders such as CF, PCD, NTM and others. Therefore, physicians should be familiar with the key clinical history and CT features that can raise the suspicion of a specific cause of bronchiectasis and lead to improved treatment.
Footnotes
Provenance: Commissioned article, peer reviewed.
Conflict of interest: J.D. Chalmers reports grants and personal fees (for COPD) from GlaxoSmithKline, Boehringer Ingelheim and Pfizer, grants (for COPD) from AstraZeneca, grants and personal fees (for research into bronchiectasis) from Bayer Healthcare and Grifols, and personal fees (for consultancy) from Napp, outside the submitted work.
- Received February 23, 2018.
- Accepted June 4, 2018.
- Copyright ©ERS 2018.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










