





Abstract
Fibrotic interstitial pneumonias are a group of rare diseases characterised by distortion of lung interstitium. Patients with mutations in surfactant-processing genes, such as surfactant protein C (SFTPC), surfactant protein A1 and A2 (SFTPA1 and A2), ATP binding cassette A3 (ABCA3) and Hermansky–Pudlak syndrome (HPS1, 2 and 4), develop progressive pulmonary fibrosis, often culminating in fatal respiratory insufficiency. Although many mutations have been described, little is known about the optimal treatment strategy for fibrotic interstitial pneumonia patients with surfactant-processing mutations.
We performed a systematic literature review of studies that described a drug effect in patients, cell or mouse models with a surfactant-processing mutation. In total, 73 articles were selected, consisting of 55 interstitial lung disease case reports/series, two clinical trials and 16 cell or mouse studies. Clinical effect parameters included lung function, radiological characteristics and clinical symptoms, while experimental outcome parameters included chemokine/cytokine expression, surfactant trafficking, necrosis and apoptosis. SP600125, a c-jun N-terminal kinase (JNK) inhibitor, hydroxychloroquine and 4-phenylbutyric acid were most frequently studied in disease models and lead to variable outcomes, suggesting that outcome is mutation dependent.
This systematic review summarises effect parameters for future studies on surfactant-processing disorders in disease models and provides directions for future trials in affected patients.
Abstract
Drug effects in disease models of surfactant-processing disease are highly dependent on mutation http://ow.ly/ZYZH30k3RkK
Introduction
Idiopathic interstitial pneumonias (IIPs) are a rare group of diseases characterised by distortion of lung interstitium. IIPs can be subclassified into fibrotic interstitial pneumonia (FIP), smoking-related interstitial pneumonia and acute/subacute interstitial pneumonia [1]. The aetiology of IIPs is unknown; however, affected patients commonly have a first-degree relative with pulmonary fibrosis, referred to as familial interstitial pneumonia. It has been suggested that up to 20% of FIP cases might be familial [2, 3]. In FIP, two distinct groups of causal genetic mutations have been recognised; surfactant-processing and telomere maintenance gene mutations. To date, mutations in four surfactant-associated genes have been found to cause pulmonary fibrosis: surfactant protein C (SFTPC) [4, 5], surfactant protein A1 (SFTPA1) [6], surfactant protein A2 (SFTPA2) [7, 8] and ATP binding cassette transporter (ABCA3) [9, 10]. Furthermore, mutations in Hermansky–Pudlak syndrome 1 (HPS1) and 4 (HPS4) can cause pulmonary fibrosis in patients with Hermansky–Pudlak syndrome (HPS) with a lung phenotype equalling that of FIP [11, 12]. In addition, it has been found that Hermansky–Pudlak syndrome 2 (HPS2, associated with mutations in gene AP3B1) can cause pulmonary fibrosis in children and only results in mild interstitial lung disease (ILD) in adults [13]. SFTPC, SFTPA1 and SFTPA2 encode surfactant proteins (SPs)-C, -A1 and -A2, respectively, that have various biophysical functions and protect alveoli against damage and infection [14, 15]. The genes ABCA3, HPS1, AP3B1 and HPS4 encode structural proteins of lamellar bodies, the characteristic organelles in alveolar type II cells (AEC2) that are crucial for surfactant processing [16–18]. In general, mutations in surfactant-processing genes seem to cause misfolding or inappropriate localisation of proproteins, which leads to accumulation of proprotein in the endoplasmic reticulum or in inappropriate cellular compartments or in degradation of the proprotein [19]. In turn, this results in altered cellular processes in AEC2, such as dysregulated proteostasis, altered surfactant lipid composition and activation of immune cells in SFTPC non-BRICHOS mutations [20, 21], endoplasmic reticulum stress in SFTPC BRICHOS mutations and SFTPA1 and 2 mutations [22–26] and impaired lipid transport, dysfunctional lysosome-related organelles, increased endoplasmic reticulum stress and apoptotic signalling in ABCA3 and HPS mutations [17, 27–29]. In addition to the above disease-causing mutations, single nucleotide polymorphisms in the MUC5B and TOLLIP genes have been associated with predisposition to idiopathic pulmonary fibrosis (IPF), as well as survival in IPF patients [30–32]. Recent studies have identified various molecular phenotypes in IPF patients. These different molecular phenotypes correspond to variance in disease behaviour, and possibly to the response to different treatment regimens. Stratification of FIP patients based on genetic characteristics as well as cellular and molecular biomarkers could lead to personalised treatment strategies in the future [33, 34]. However, previous therapeutic trials have mostly not tested what genetic characteristics and biomarkers are associated with a good treatment response. Therefore, it is not known whether FIP patients with surfactant-processing mutations should receive the same therapy as other FIP patients. They might benefit from a different treatment strategy.
The aim of this systematic review is to provide an overview of studies that investigated drug effects in patients, cell or mouse models containing a mutation in surfactant-processing genes involved in pulmonary fibrosis. This review will focus on drug types, effect parameters and outcomes. This will provide a basis for future research efforts into treatment strategies for FIP patients with surfactant-processing mutations.
Materials and methods
Data sources and literature searches
A literature search in the electronic databases of Pubmed and Embase was performed with the help of a clinical librarian. We selected studies that contained, in the Medical Subject Headings (MeSH), keywords or text words, at least one search item from each of the following three groups: 1) ILD, with MeSH terms referring to ILD or lung cells; 2) surfactant-processing mutation, with MeSH terms referring to genes involved in adult FIP, pulmonary surfactant-associated protein, Hermansky–Pudlak syndrome or ATP-binding cassette transporter; 3) treatment, with MeSH terms referring to drug, treatment or therapy. The search was restricted to articles written in English and published before July 2, 2017. In the Embase search, conference abstracts were excluded. To maximise the inclusion of case reports/series, a second search was performed. This search included search items from groups 1 and 2, but not group 3 (treatment). This second search was restricted to the categories case report, clinical article, clinical study, clinical trial (phase I–IV), (classical) article, cohort analysis, comparative study, controlled study, controlled clinical trial, human, human tissue, major clinical study, retrospective study, evaluation study, letter, multicentre study, observational study, pragmatic clinical trial and randomised control trial. Duplicates within the search with two search items and the search with three search items were identified and removed using the reference management programme RefWorks (Ann Arbor, MI, USA). Duplicates between the two searches were removed manually. The complete search strategy is provided in the online supplementary material.
Study selection
Title and abstract of the retrieved articles were reviewed and articles were scored based on the following three criteria. Studies involving 1) ILD, mouse models or cells and lung disease; 2) surfactant-processing mutation involved in adult pulmonary fibrosis; and 3) original research articles. Abstracts that were scored for all three criteria were selected and the full-text versions were reviewed. Studies were excluded that reported no or only non-pharmacological drug effects, such as small interfering RNA/short hairpin RNA, gene overexpression, supplemental oxygen, bronchoalveolar lavages and lung transplantation. Case reports/series were excluded in which HPS diagnosis was not based on genetic analysis or absence of dense granules in platelets assessed by electron microscopy. The references of the finally selected articles were screened for additional eligible studies.
Classification of drug effect in case reports/series
Drug effect in case reports or series of paediatric and adult cases was determined based on the information found in journal articles. Different terms, such as improvement, short-term improvement, stabilisation, short-term stabilisation, limited effect or no effect were used to express outcome of treatment. Sick-better, (some) improvement of lung function, clinical or respiratory symptoms, chest film or high-resolution computed tomography (HRCT) or just improvement after treatment was defined as improvement; transient effect or improvement and later a reduced effect of treatment or deterioration of disease was defined as short-term improvement; stable condition, stabilisation of lung function, clinical symptoms or sick-same was defined as stabilisation. Short-term stabilisation was used when this drug effect was only present for a short period of time. No effect after treatment, deterioration of lung function, clinical symptoms or HRCT was defined as no effect; and limited effect was used when little effect or minimal improvement followed by death was reported.
Results
Search results
In figure 1, the flow diagram of the search and study selection process is displayed. Two different searches, one with three groups of search terms (ILD/lung cells, surfactant-processing mutation and treatment) and one with two groups of search terms (ILD/lung cells and surfactant-processing mutation) were performed. The searches resulted in a total of 1878 unique articles. The full text was read of 239 articles, of which 73 were selected to be included in this review. Selected studies consisted of 16 studies performed in cell or mouse models, 55 case reports/series and two clinical trials. Mutations in SFTPC, HPS1 and ABCA3 were most frequently studied. The number of case reports/series, clinical trials and cell/mouse studies per mutation are displayed in table 1.

Flowchart of the article selection process. HPS: Hermansky–Pudlak syndrome.
Included studies on surfactant-processing mutations categorised by study type
Drugs and effect parameters used in case reports/series and cell/mouse studies
The studied drugs can be divided into immunosuppressive agents, antifibrotic agents, mitogen-activated protein kinase signalling pathway inhibitors, antibiotics, combination therapy, anti-apoptotic therapy and other therapies. 55 case reports/series were included, of which 43 described paediatric cases, nine described adult cases and three described paediatric as well as adult cases. Adult patients were treated with corticosteroids, cyclosporine A, antibiotics, pirfenidone and/or azathioprine. Additional drugs used in paediatric patients were exogenous surfactant and hydroxychloroquine. Two clinical trials were included in which patients were treated with pirfenidone (table 2). The same drugs were tested in cell and mouse models (table 3). Additionally, drugs tested in cell and mouse models were glycerol, rapamycin, 4-phenylbutyric acid (PBA), saralasin, angiotensin (ANG)1–7, cyclophosphamide, recombinant CHI3L1, interleukin (IL)13Rα2 construct, antibodies against monocyte chemotactic protein (MCP)-1 or SP-D and specific inhibitors for c-Jun N-terminal kinases (JNK), caspase 4, ADAM metallopeptidase domain 17/tumor necrosis factor-α-converting enzyme (ADAM17/TACE), synoviolin and extracellular signal-regulated kinases (ERK1/2, P38, nuclear factor (NF)-κB and CRTH2).
Case reports/series and clinical trials of humans with a surfactant-processing mutation involved in fibrotic interstitial pneumonia
Drug effect in cell and mouse models with a surfactant-processing mutation
Human studies reported clinical symptoms, lung function, radiological characteristics or oxygen requirement as effect parameters. A majority of the cell-line and mouse studies investigated alterations in processes involved in surfactant trafficking [21, 35, 56–58], cytokine/chemokine concentrations [59, 60, 86, 99] and necrosis/apoptosis [21, 57, 61–63, 100], while some studies investigated weight loss, airway compliance [60], collagen secretion/accumulation [64, 100], migration of macrophages [101] or mortality [102] (table 3 and figure 2).

{kind=link}
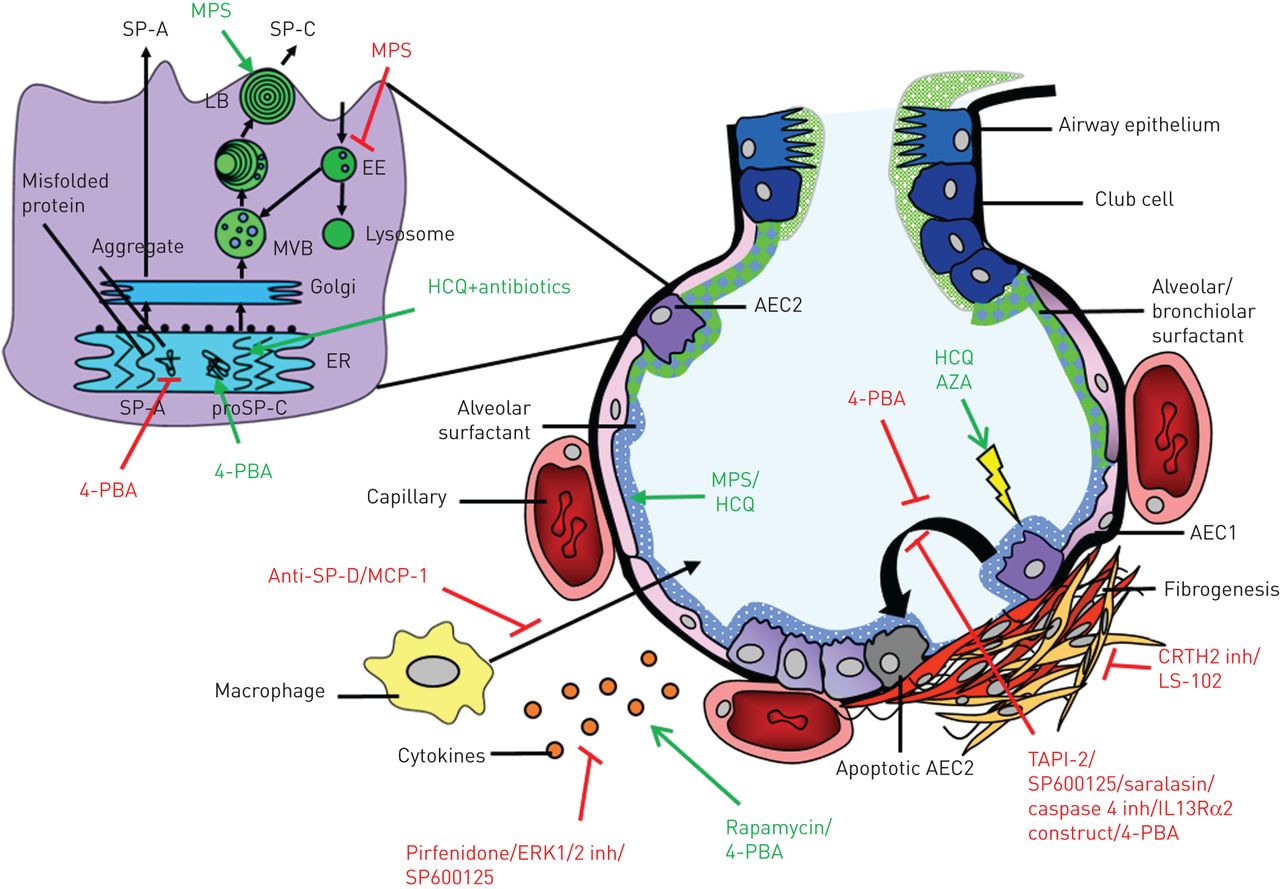{kind=link}
Targets of drugs investigated in humans and disease models with a surfactant-processing mutation. Damage to alveolar tissue causes fibrogenesis, which can be targeted by drugs in multiple ways. Expanded section: alveolar type II cell (AEC2) with organelles involved in surfactant processing. MPS: methylprednisolone; SP: surfactant protein; LB: lamellar bodies; EE: early endosomes; MVB: multivesicular bodies; ER: endoplasmic reticulum; 4-PBA: 4-phenylbutyric acid; MCP: monocyte chemotactic protein; ERK: extracellular signal-regulated kinases; HCQ: hydroxychloroquine; AZA: azathioprine; AEC1: alveolar type I cell; inh: inhibitor.
Outcome in case reports/series and clinical trials
An extensive description of the effect of treatment in paediatric and adult cases with a surfactant-processing mutation is described in online supplementary table S1. A concise outcome after treatment per drug combination (no effect, (short-term) improvement, (short-term) stabilisation, little effect) was determined based on the information derived from case reports/series. In addition, an overall outcome after treatment was determined based on the last reported outcome after treatment with different drugs and displayed in table 2. In six of the 12 adult case studies (five patients in total) [7, 10, 85, 89–91], (short-term) improvement or (short-term) stabilisation of the disease was observed after treatment with pirfenidone, corticosteroids or antibiotics. In paediatric case studies (short-term) stabilisation or (short-term) improvement of disease was observed in 20 of the 23 case reports/series [5, 36–51, 87, 88, 103] (in five case series, not in all described cases; total 67 patients) describing patients with a SFTPC mutation treated with hydroxychloroquine, surfactant, antibiotics or corticosteroids. In addition, (short-term) improvement or (short-term) stabilisation of disease was found in 15 of the 24 case reports/series describing paediatric cases [10, 65–76, 85, 88] (in four case series, not in all described cases; 28 patients in total) describing patients with an ABCA3 mutation treated with the same drugs. In one case report describing a drug effect of corticosteroids in a patient with an AP3B1 mutation, stabilisation of disease was reported [13]. One of the two clinical trials included in this review [97] reported a positive effect of pirfenidone on lung function of HPS patients with a baseline forced vital capacity (FVC) ≥50% predicted. The other clinical trial reported no statistically significant difference between pirfenidone and the placebo group [98].
Outcome in mouse studies and cells derived from humans with an HPS1 mutation
The effect of drugs on mouse or human lung cells with an HPS1 mutation was investigated in three studies [99–101]. Treatment of alveolar macrophages from bronchoalveolar lavage (BAL) fluid derived from patients with an HPS1 mutation with pirfenidone resulted in reduced cytokine/chemokine secretion [99]. In addition, changes in macrophage behaviour were found in experiments with BAL fluid from patients with an HPS1 mutation and Hps1ep/Hps1ep, Ap3b1pe/Ap3b1pe (EPPE) C57BL/6J mice treated with anti-MCP1 and/or anti-SP-D. This resulted in reduced macrophage migration [101]. In addition, bleomycin-treated HPS1 mutated mouse AEC2 were treated with CHI3L1, IL13Rα2 and CRTH2, which resulted in no effect on apoptosis, amelioration of apoptosis and reduced collagen accumulation, respectively [100].
Outcome in cell lines with a pulmonary surfactant associated mutation
The most frequently studied drugs in cell lines with a surfactant-processing mutation are 4-phenylbutyric acid (n=5), JNK inhibitor (n=3), hydroxychloroquine (n=3), methylprednisolone (n=2), azathioprine (n=2) and cyclophosphamide (n=2). Hydroxychloroquine or methylprednisolone treatment was found to have a positive effect on lysophosphatidylcholine and phosphatidylcholine levels in MLE12 cells transfected with SFTPCI73T [21] or SFTPCA116D [57]. In addition, treatment with methylprednisolone, but not hydroxychloroquine, resulted in partial correction of the mislocalisation of pro-SP-C in MLE12 cells expressing SP-CI73T [21]. Furthermore, another study with MLE12 cells expressing SP-CL188Q treatment with hydroxychloroquine resulted in increased accumulation of pro-SP-C [56]. Azathioprine seems to have a negative effect on MLE12 cells transfected with SFTPCI73T or SFTPCA116D, as evidenced by increased LDH levels after treatment. For cyclophosphamide treatment only an effect on chaperone protein expression of heat shock protein (HSP)70 and HSP90 could be found [21, 57].
In A549 cells transfected with SFTPCΔexon4, it was found that 4-PBA attenuated increased NF-κB expression, which is a marker for cellular stress response. However, it had no inhibitory effect on pro-inflammatory interleukin (IL)-8 production [59]. In addition, 4-PBA resulted in reduced NP-40 insoluble aggregate formation and increased protein secretion of SP-A2G231V and SP-A2F198S expressing CHO-K1 cells [35] and on juxtanuclear accumulation of pro-SPC in SFTPCΔexon4-mutated A549 cells [58]. In contrast, 4-PBA resulted in a slightly increased accumulation of SP-CΔexon4 and SP-CL188Q proprotein in transfected HEK293 cells. Furthermore, treatment of HEK293 cells transfected with SFTPCL188Q with 4-PBA increased endoplasmic reticulum stress and accumulation of detergent insoluble aggregates, but it also resulted in increased mature SP-CI73T and SP-CL188Q protein [56]. In contrast, Nguyen and Uhal [63] showed that treatment of A549 cells transfected with SFTPCG100S with 4-PBA resulted in attenuation of increased endoplasmic reticulum stress-induced factors. This study also showed that treatment with 4-PBA can result in reduced nuclear fragmentation.
Another frequently studied drug in cell models is the JNK inhibitor SP600125, sometimes in combination with 4-PBA or a caspase-4 inhibitor. In A549 cells transfected with SFTPCΔexon4, this drug resulted in reduced IL-8 concentration [59] and DNA fragmentation [61], although it had no effect on IL-8 concentration in A549 cells transfected with ABCA3T1173R [86]. Other drugs not currently used in ILD, e.g. synoviolin inhibitor and saralasin were tested in cell lines with a surfactant-processing mutation by assessing collagen secretion and endoplasmic reticulum-stress induced processes such as apoptosis or expression of chemokines and cytokines. These drugs were only tested in one study, and yielded both positive and negative results.
Discussion
This systematic review provides an overview of studies that investigated the effect of drugs on patients with ILD and a surfactant-processing mutation or cell or mouse models with a surfactant-processing mutation involved in pulmonary fibrosis. Human studies reported only treatment with antibiotics and drugs against inflammatory or fibrotic processes and evaluated lung function and radiological characteristics over time. Although some positive results were reported in adult case reports/series, except for one case report [10] no curative or long-term stabilising effects on pulmonary fibrosis were reported. In more than half of the case reports/series of children, stabilisation or improvement of disease after treatment was reported. Cell and mouse studies used drugs that interfered with aberrant biological processes related to surfactant mutations. This heterogeneous group of studies showed that results are gene- and mutation-dependent and yielded results that may contribute to the development of personalised medicine in the future.
In two cell line studies with SFTPCI73T- and SFTPCA116D-mutated cells, addition of methylprednisolone showed the most promising results as it partially corrected mislocalisation of SFTPC and (partially) corrected altered (lyso)phospholipid levels [21, 57]. In addition, glucocorticosteroids have been used for the treatment of patients; in three adult cases [7, 10, 89] with a surfactant-processing mutation, improvement or minimal progression of disease was observed after treatment with prednisone. Two of these patients had diseases that are known to often respond to immunosuppressive therapy. One had concomitant pulmonary sarcoidosis [89] and one had hypersensitivity pneumonitis [7]. Two other adult cases [90, 91] treated with corticosteroids and pirfenidone showed (short-term) stabilisation of the disease. In paediatric cases described in case reports/series included in this review, treatment with antibiotics, hydroxychloroquine or corticosteroids resulted more often in (short-term) improvement or (short-term) stabilisation of disease compared to adult cases. The difference in response may be due to the difference in clinical phenotype between adult and paediatric patients with surfactant-processing mutations. Children commonly present with desquamative interstitial pneumonia, non-specific interstitial pneumonia, pulmonary alveolar proteinosis, chronic pneumonitis of infancy or respiratory distress syndrome, but seldom with IPF. In addition, host environment, initial injury and regenerative capacity of tissue [104] may differ between adults and children.
It is difficult to draw conclusions only based on case reports/series, since the clinical parameters reported are limited and the information is often not quantitative. In addition, only interesting cases are selected for a case report, introducing bias into the results. Griese and colleagues have initiated a trial to investigate the effect of hydroxychloroquine in paediatric ILD in a more standardised way (ClinicalTrials.gov identifier NCT02615938). A significant subgroup of the patients is expected to carry surfactant-processing mutations; therefore, the results of this trial will provide evidence for therapeutic intervention in paediatric patients with these mutations. However, based on the difference between paediatric and adult patients carrying similar surfactant-processing mutations, translation of these results to clinical management of adult patients will need to proceed with utmost care. In addition, immunosuppressive treatment of adult patients with FIP was shown to be unfavourable in patients with IPF. A negative effect on survival and hospitalisation was found in IPF patients receiving the combination treatment of prednisone, azathioprine and N-acetylcysteine in the PANTHER-IPF trial [105] and recently the negative effect of glucocorticoid treatment was reported for a retrospective cohort of suspected IPF patients [106].
Most reports on patients with a surfactant-processing mutation show that disease progresses as monitored by change deterioration of FVC. Clinical trials were only conducted in HPS patients. In two clinical trials the drug pirfenidone was tested. One trial described a positive effect of pirfenidone on slowing down lung function decrease [97], whereas the other clinical trial found no statistically significant difference between the placebo and pirfenidone group [98]. The placebo group of the last clinical trial showed a small rate of decline in FVC. The positive result of the first trial was closely comparable to that reported by King et al. [107] who showed that pirfenidone is effective in slowing down FVC decrease in a cohort of IPF patients (with unknown genetic characteristics) with a baseline FVC ≥50% predicted. The results of both trials included in this review do not provide unambiguous evidence on whether pirfenidone would be helpful for FIP patients with HPS mutations.
In model systems drugs were tested that intervened with the aberrant processes directly related to the surfactant-processing mutation, such as surfactant trafficking, cytokine/chemokine expression, necrosis and apoptosis. The most frequently studied drug in the cell and mouse model studies included in this review is 4-PBA. 4-PBA, a hydrophobic chemical chaperone with a role in promoting trafficking of misfolded proteins, has been approved by the United States Food and Drug Administration for treatment of urea cycle disorders. In addition, its therapeutic effects on other pathologies, such as neurological diseases, diabetes type 2 and protein folding diseases are now being investigated (reviewed by Kolb et al. [108]). For 4-PBA a positive effect, and, with other mutations, a negative effect on aggregate formation [35, 56], accumulation of SP-C [56, 58] and SP-C mature protein expression [35, 56] in different SP-A2- and SP-C-mutated cells has been described. In addition, one study has shown that 4-PBA treatment can reduce nuclear fragmentation in SFTPCG100S lung cells [63]. Therefore, further studies are needed to provide evidence for a possible role of 4-PBA in the treatment of patients with surfactant-processing mutations. Other agents, inhibitors against synoviolin, CRTH2, JNK and ANGII receptors showed interesting results in cell studies with a surfactant-processing mutation by reducing collagen secretion/accumulation or nuclear fragmentation. However, future studies, such as replication of cell studies and research in mouse models, including monitoring of side-effects, are still needed.
The investigation of drugs for patients with a SFTPC mutation is complicated by the fact that each pro-SP-C mutation seems to result in unique effects on intracellular trafficking of pro-SP-C and the presence of the mature form of SP-C. Even between mutations that are in the same functional domain of the protein (BRICHOS domain) different effects on SP-C processing have been observed [109]. For example, HEK293 cells transfected with SFTPCI73T showed increased accumulation of pro-SPC compared to wild-type, whereas SFTPCL188Q and SFTPCΔexon4 showed reduced accumulation of pro-SPC [56].
In summary, different drugs have been tested in different cell lines with a surfactant-processing mutation using different outcome measures. Many of these experiments have only been performed once. To investigate the effect of surfactant-processing mutations and the effect of drugs, the development of a new model for IIPs that better represents affected human lungs is highly wanted. In future, lung organoids, in vitro three-dimensional lung cell models, may fill the gap between cell lines and humans. Tracheobronchial organoids [110] have already been generated from human tissue explants or biopsies. Stable distal lung organoids, which would be necessary to model surfactant processing adequately, have only been generated from tissue derived from mice [111, 112]. Previously generated distal lung organoids from human lung had a low viability [113], no turnover [114] or high transdifferentiation to type I alveolar epithelial cells [115]. Interestingly, in organoids generated from human-induced pluripotent stem (iPS) cells, alveolar structures have been observed [116, 117]. However, it must be remembered that iPS cells retain characteristics of their cell of origin [118], which might influence their drug response.
In conclusion, this review shows promising drugs described in case reports/series, clinical trials and disease models. One of the two trials in patients with HPS show that patients with surfactant-processing mutations might benefit from anti-fibrotic drugs. Cell and mouse models show that interference with mutation-dependent aberrant processes yield positive results. However, the results seem to be highly gene- and mutation-specific. Translation of these results into personalised medicine is not possible at present. Hopefully, the development of new disease model systems with appropriate outcome parameters will make it possible to test drugs on human lung cells with a specific introduced or native surfactant-processing mutation [119], leading to improved treatment strategies for patients with FIP.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
online supplement ERR-0135-2017_online_supplement
Footnotes
This article has supplementary material available from err.ersjournals.com
Provenance: Submitted article, peer reviewed.
Conflict of interest: None declared.
- Received December 18, 2017.
- Accepted April 12, 2018.
- Copyright ©ERS 2018.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










