





Abstract
Classifying pulmonary fibrotic disease into various diagnostic categories provides the clinician with expectations for both prognosis and proper treatment. Despite years of experience with histological, radiological and clinical guidelines, a group of patients remains with unclassifiable interstitial lung disease. In this article, the possible barriers to classification will be explored, and some strategies will be discussed to aid in overcoming these barriers.
Abstract
What are the sources of unclassifiable interstitial fibrosis? What factors prevent a definitive diagnosis? http://ow.ly/PMY530iUCCY
Introduction
For over a half-century, histological examination of lung biopsies has provided information that allows for both prognoses and treatment decisions in patients with lung disease. Although tabular lists of diseases existed prior to 1960 [1], papers by Averill Liebow and Charles Carrington in the late 1960's provided the first histopathological framework for classification of interstitial lung diseases [2, 3]. This system was originally billed as a classification of idiopathic interstitial pneumonias; however, over the years, several of the described entities were shown to be related to various exposures or diseases. For example, nearly all cases of giant-cell interstitial pneumonia are now classified as hard metal pneumoconiosis and are known to be secondary to exposure to tungsten or cobalt, desquamative interstitial pneumonia is most commonly associated with smoking of cigarettes, and lymphocytic interstitial pneumonia is frequently secondary to autoimmune connective tissue disease or immunodeficiency. In addition, the entity described as usual interstitial pneumonia originally covered a fairly broad group of diseases that included those now classified as nonspecific interstitial pneumonia as well as usual interstitial pneumonia [4]. The current American Thoracic Society (ATS)/European Respiratory Society (ERS) classification system has remedied some of these problems, acknowledging the known causes of many of the patterns and splitting some diseases out of the classification scheme entirely [5]. However, the inability of the system to address overlapping or complex histology results in occasional cases eluding categorisation.
There is a saying in pathology that there are two types of pathologists: honest pathologists and famous pathologists. The semblance of omniscience has traditionally been a prerequisite for obtaining the mantle of “expert”. The facade of this dogma was thankfully partially torn down in the 2002 ATS/ERS classification when the diagnostic category “unclassifiable interstitial fibrosis” was proposed [6]. This statement that not all cases could be categorised into one of the defined idiopathic interstitial pneumonias helped establish a label for those biopsies that were previously either forced into a defined category or were more honestly referred to as “difficult to classify” [7].
In the pulmonary literature, unclassifiable interstitial lung disease refers to cases that defy classification following input from several medical specialties [8]. This combination of clinical, radiological and pathological data is most commonly referred to as multidisciplinary discussion (MDD). In various studies, the percentage of unclassifiable cases to total patients is approximately 10–25% [8–11]. The inability to accurately classify cases is usually the result of the following: 1) a biopsy was not performed (and clinical and radiological data were insufficient for accurate diagnosis); 2) there were discrepant or overlapping histopathological findings; 3) there were discrepant features between clinical, radiological and pathological findings; and 4) there were significant overlapping clinical or radiological features that precluded diagnosis.
Surgical lung biopsy: reducing pre-histological barriers to classification
The decision to perform a surgical biopsy depends upon the perceived benefit derived from histopathologic examination balanced against the perceived mortality and morbidity of the procedure [12]. The risk of mortality has been recently evaluated [13, 14]. In two large series of surgical lung biopsies in the USA and UK, the 30-day mortality rate was 1.5% and 1.0% for elective surgeries, respectively. Despite this relatively low risk, there are certain subpopulations of patients that may have a higher chance of a poor outcome. There is an increased risk of mortality in patients with autoimmune connective tissue disease, in patients being treated with corticosteroids and in older patients.
Once a decision to perform a biopsy has been made, it is important to work intelligently to obtain a diagnostic sample. It is frustrating to the multidisciplinary team (MDT) and a disservice to the patient to perform surgery only to acquire a minute fragment of scarred pleura. Although we think about the MDT as pulmonologist, radiologist and pathologist; at the time of biopsy the surgeon and anaesthesiologist are the principal players. Prior to the biopsy, the expectations should be discussed. The surgeon, pulmonologist and radiologist determine the best foci for sampling. In our practice, we try and obtain tissue from two to three lobes, focusing on regions with active disease (such as interfaces between fibrotic and less-involved regions), and attempt to procure tissue up to a depth of 1.5–2 cm from the pleural surface (which usually results in a biopsy with a diameter of approximately 4–5 cm). Specimens should be labelled with the position within the lobe (e.g. posterior–basilar lower lobe). During the surgery, the anaesthesiologist should be mindful of minimising stretch injury by using as low a tidal volume as possible. This is thought to decrease likelihood of post-operative acute exacerbation [15, 16]. The anaesthesiologist should also use as low a fraction of inhaled oxygen as possible to minimise the risk of oxidant-induced injury.
Typical patterns of fibrosis
It is useful to consider the pattern of fibrosis relative to its distribution within the pulmonary lobule. When viewed in this manner, there are three typical patterns of fibrosis encountered in lung biopsy samples: bronchiolocentric fibrosis, nonspecific interstitial pneumonia fibrosis and usual interstitial pneumonia.
Bronchiolocentric fibrosis is characterised by thickening of peribronchiolar alveolar septa by collagen deposition (figure 1a). This is often accompanied by extension of the bronchiolar epithelium onto these thickened septa, so-called peribronchiolar metaplasia (figure 1b).

Bronchiolocentric fibrosis. a) This low power view shows fibrosis of peribronchiolar alveolar septa. Scale bar=500 µm. b) A higher power view shows peribronchiolar metaplasia with lining of the bronchiolocentric alveolar septa by ciliated columnar respiratory epithelium. Scale bar=200 µm.
Nonspecific interstitial pneumonia is characterised by diffuse alveolar septal thickening by collagen deposition such that the peribronchiolar, subpleural and alveolar septa between these regions all show similar abnormalities (figure 2a). This pattern of fibrosis maintains the underlying pulmonary alveolar architecture resulting in a “dusty cobweb” appearance (figure 2b).

Nonspecific interstitial pneumonia. a) This low power view shows diffuse alveolar septal thickening by moderate interstitial fibrosis. Scale bar=1 mm. b) A higher power view shows the architectural preservation of lung tissue. Scale bar=500 µm.
Usual interstitial pneumonia is characterised by fibrosis that is accentuated at the periphery of the pulmonary lobule, in the subpleural regions and adjacent to interlobular septa. The subpleural regions often show microscopic honeycombing with irregular airspaces lined by bronchial epithelium, surrounded by dense fibrosis (figure 3a). The peribronchiolar alveoli are thin and delicate, and there are often fibroblast foci at the interface between fibrotic and less-involved regions. These fibroblast foci are bulge-like regions adjacent to interstitial scarring and are composed of bland fibroblasts arranged parallel to the airspace, within a myxoid matrix, with a reactive cap of cuboidal epithelial cells (figure 3b). The cystic spaces of advanced bronchiolocentric fibrosis may mimic microscopic honeycombing. However, in typical cases of usual interstitial pneumonia, the largest cysts are in a subpleural distribution while in bronchiolocentric fibrosis, the largest cysts are often separated from the pleura by smaller airspaces from residual fibrotic alveoli. Organising pneumonia, particularly when resolving, may mimic fibroblast foci. However, the pathologist should look for characteristic features of rounded branching polypoid plugs of oedematous fibromyxoid tissue, often surrounded by macrophages or air rather than the flattened elongate region of fibroplasia with adjacent dense fibrosis typical of fibroblast foci.

Usual interstitial pneumonia. a) This low power view shows the heterogeneous pattern of fibrosis with subpleural microscopic honeycombing and centrilobular lack of alveolar inflammation or fibrosis. Scale bar=2 mm. b) A high power view shows a fibroblast focus with typical layered appearance within a loose myxoid stroma. Scale bar=100 µm.
Physiological basis of the typical patterns of fibrosis
The three patterns of fibrosis evoke three different physiological mechanisms, the knowledge of which aid in constructing a differential diagnosis. These three corresponding mechanisms of disease are inhalation injury or airway inflammation, alveolar inflammation and abnormal senescence.
Bronchiolocentric fibrosis is most commonly the result of centrilobular injury due to either fume or dust inhalation, aspiration or from systemic disease manifesting with airway inflammation. Classical diseases with bronchiolocentric fibrosis include respiratory bronchiolitis from smoking, hypersensitivity pneumonia from immune reaction to inhaled antigens and asbestosis from inhalation of asbestos fibres. Systemic diseases that result in bronchiolocentric fibrosis include Sjogren syndrome and inflammatory bowel disease-related small airway disease.
Nonspecific interstitial pneumonia-fibrosis is most commonly the result of diffuse alveolar inflammation followed by fibrosis. This pattern of inflammation is characteristic of autoimmune connective tissue disease, drug reactions, some cases of hypersensitivity pneumonia and the organising phase of diffuse alveolar damage. These diseases are often driven by circulating factors that affect the alveolar tissue more diffusely.
Usual interstitial pneumonia is a peculiar pattern of fibrosis and several mechanisms have been proposed historically including alveolitis and aberrant healing response [17]. Currently, a major component of this disease is believed to arise from aberrant senescence. Several series of idiopathic pulmonary fibrosis have shown that patients with idiopathic pulmonary fibrosis have significantly shorter telomeres than matched controls and have more frequent mutations in the telomerase pathway (e.g. telomerase RNA component (TERC) and telomerase reverse transcriptase (TERT)) [18–21]. It has been proposed that alveolar epithelial cells are derived from bronchioloalveolar stem cells located in the terminal bronchioles and from multipotent type 2 pneumocytes in distal lung [22]. This results in the oldest type 1 pneumocytes being located in the distal peripheral acinar tissues (i.e. in the subpleural and distal paraseptal alveoli). In addition, these distal alveolar units are subjected to increased tractional stress during normal inspiration and coughing. While the above explanation is predominantly speculative, it provides a rationale for the distribution of the abnormal fibrosis in usual interstitial fibrosis.
It is possible that the aforementioned third physiological mechanism results in a susceptibility to lung fibrosis that may be propelled either by accelerated senescence (resulting in typical usual interstitial pneumonia) or by the addition of either of the first two mechanisms (resulting in a mixed pattern of fibrosis). These mixed patterns of fibrosis may result in biopsy samples that are difficult to classify. An intriguing project for future study would be evaluation of unclassifiable interstitial fibrosis cases for evidence of similar mechanisms of abnormal senescence.
Histological sources of unclassifiable interstitial fibrosis
Several histological reasons for a diagnosis of unclassifiable interstitial fibrosis exist: 1) the biopsy is too small; 2) the biopsy shows only advanced interstitial fibrosis; 3) the histology is altered by pre-biopsy treatment; and 4) there are overlapping features that do not allow classification into a pure category.
The too-small biopsy
Unfortunately, solving the problem of the too-small biopsy is nearly impossible once the biopsy has been performed. As mentioned above, there are interventions that can be used in order to minimise the likelihood of obtaining a non-diagnostic biopsy due to sampling error. Once the biopsy is obtained, there are methods to maximise the ability to make a definitive diagnosis. These are basically techniques of properly fixing and cutting the biopsy. The staple line should be excised as tightly as possible without sacrificing any additional biopsy tissue. After the staples have been removed, there are two typical methods of fixation. If the biopsy appears to have been properly handled (not excessively squeezed) it can be placed in a small sealed container of formalin and shaken vigorously. This results in near complete expansion of the artefactual atelectasis that has resulted from the stapling of the lung. Alternately, a small syringe with narrow gauge needle can be used to gently inflate the sample. The biopsy should then be cut perpendicular to the pleural surface in order to have the highest likelihood of demonstrating the greatest cross-sectional diameter of complete pulmonary lobules from pleural surface to proximal airway.
In the final interpretation, there will be situations in which a biopsy will be unclassifiable due to small sample size. In these instances, it is important to discuss the morphological findings, state whether they are compatible or incompatible with the clinical and radiological features, and state that interpretation is limited by small sample size.
The biopsy with advanced fibrosis
The biopsy with only advanced fibrosis is difficult to classify into an aetiological framework since many of the interstitial pneumonias can show diffuse fibrosis as their final pattern [23]. However, broad zones of fibrosis with enlarged irregular subpleural airspaces are most commonly associated with idiopathic pulmonary fibrosis, and a biopsy that shows only microscopic honeycombing meets the ATS/ERS histopathological criteria for probable usual interstitial pneumonia [24].
These biopsies often share features of the too-small biopsy, and one should try and use the methods described above to try and maximise the amount of tissue available for evaluation. This can result in finding rare zones of normal appearing lung with intervening fibroblast foci that are helpful in rendering a diagnosis of usual interstitial pneumonia. In the absence of any pathological findings other than honeycombing, a diagnosis of advanced interstitial fibrosis with mention of the presence of microscopic honeycombing or fibroblast foci is likely most prudent.
The post-therapy biopsy
There are limited published data on the effects of treatment upon the interpretation of surgical lung biopsies. Administration of corticosteroids prior to the biopsy is thought to result in alteration of the inflammatory milieu and may affect various inflammatory cells over the course of days (eosinophils), weeks (lymphocytes) and months (macrophages and granulomas). Most of these data are the result of radiographic resolution of cases of eosinophilic pneumonia or organising pneumonia. Within histopathology, the effects of steroid treatment on eosinophilic pneumonia, desquamative interstitial pneumonia and granulomatous disease are largely anecdotal [25, 26].
The biopsy with overlapping histopathological patterns
The ATS/ERS classification system is useful for both diagnosis and generating consensus regarding a given diagnosis; however, its utility is limited for the evaluation of biopsies that don't fall into strictly defined patterns. The limitation of this rigid classification has been recognised since its inception, with Liebow [2] admitting that “the alphabetical designations may be reprehensible or even barbarous, but they serve convenience”. In cases where there are overlapping histologic features, knowledge of typical scenarios that result in overlapping patterns may be useful.
Common histological patterns with overlapping features include usual interstitial pneumonia with bronchiolocentric fibrosis, nonspecific interstitial pneumonia with organising pneumonia, usual interstitial pneumonia with nonspecific interstitial pneumonia-like changes and usual interstitial pneumonia with diffuse alveolar damage. It is possible that histological purists will not appreciate mixing usual interstitial pneumonia pattern with additional features, particularly since these are often used to exclude a diagnosis of usual interstitial pneumonia. However, as Mark Twain stated “Often, the surest way to convey misinformation is to tell the strict truth.” [27]. Ruling out idiopathic pulmonary fibrosis based on the presence of additional histological features can result in failure to recognise the classical pattern of peripheral lobular fibrosis common to diseases with abnormal senescence. This limitation ends up shackling the pathologist and decreases their utility to the MDT.
Usual interstitial pneumonia with additional bronchiolocentric fibrosis
This pattern is characterised by peripheral lobular fibrosis with additional peribronchiolar metaplasia. The pathologist in this case should raise a differential diagnosis of usual interstitial pneumonia/idiopathic pulmonary fibrosis with additional injury (such as respiratory bronchiolitis from smoking or small airway disease from aspiration or fume or dust inhalation), chronic hypersensitivity pneumonia and autoimmune connective tissue disease [28–34].
Usual interstitial pneumonia with nonspecific interstitial pneumonia-like changes
In this pattern, the bronchiolocentric normal-appearing lung tissue of usual interstitial pneumonia is replaced by the uniform alveolar septal thickening more typical of nonspecific interstitial pneumonia. While technically unclassifiable, this pattern raises a differential diagnosis of usual interstitial pneumonia/idiopathic pulmonary fibrosis and autoimmune connective tissue disease (particularly rheumatoid arthritis) [28, 31, 35].
Usual interstitial pneumonia with diffuse alveolar damage
In this pattern, the typical centrilobular lung shows changes of acute lung injury including alveolar septal and airspace oedema, type 2 cell hyperplasia and hyaline membranes. Strict criteria would categorise this as unclassifiable but the differential diagnosis should include acute exacerbation of idiopathic pulmonary fibrosis and autoimmune connective tissue disease [36–44].
Nonspecific interstitial pneumonia with organising pneumonia
This pattern is not necessarily unclassifiable. Many cases of nonspecific interstitial pneumonia also have some degree of organising pneumonia. This pattern of lung injury has the same differential diagnosis as nonspecific interstitial pneumonia fibrosis itself and includes autoimmune connective tissue disease (particularly myositis syndromes and anti-synthetase syndromes), hypersensitivity pneumonia, drug reaction and idiopathic nonspecific interstitial pneumonia fibrosis [45–47].
Airspace enlargement with fibrosis/smoking-related interstitial fibrosis
The histological changes from smoking can pose a particular challenge to the pathologist. Airspace enlargement with fibrosis, also called irregular emphysema, may produce subtle subpleural scarring that is difficult to separate from early changes of usual interstitial pneumonia [48]. Histological clues to smoking-related injury include hyalinised alveolar septal fibrosis, stellate centrilobular scars and accumulation of lightly pigmented smoker’s macrophages [49]. Hyperplasia of bronchial-associated lymphoid tissue may be present. Often one will see overlapping features of emphysema and usual interstitial pneumonia, likely a consequence of the increased risk of idiopathic pulmonary fibrosis in smokers [50].
The unclassifiable biopsy: interstitial fibrosis, difficult to classify or unclassifiable pattern
Occasionally there will be a biopsy that evades all attempts at classification. These are often cases with irregular septal thickening and airspace enlargement in which differentiation between the various patterns are blurred (figures 4 and 5). In our practice, these cases represent around 5% of the total number of surgical biopsies. Approximately half of these cases can be further categorised based on clinical and radiological data; however, the other half end up with the multidisciplinary designation of unclassified interstitial lung disease (table 1).

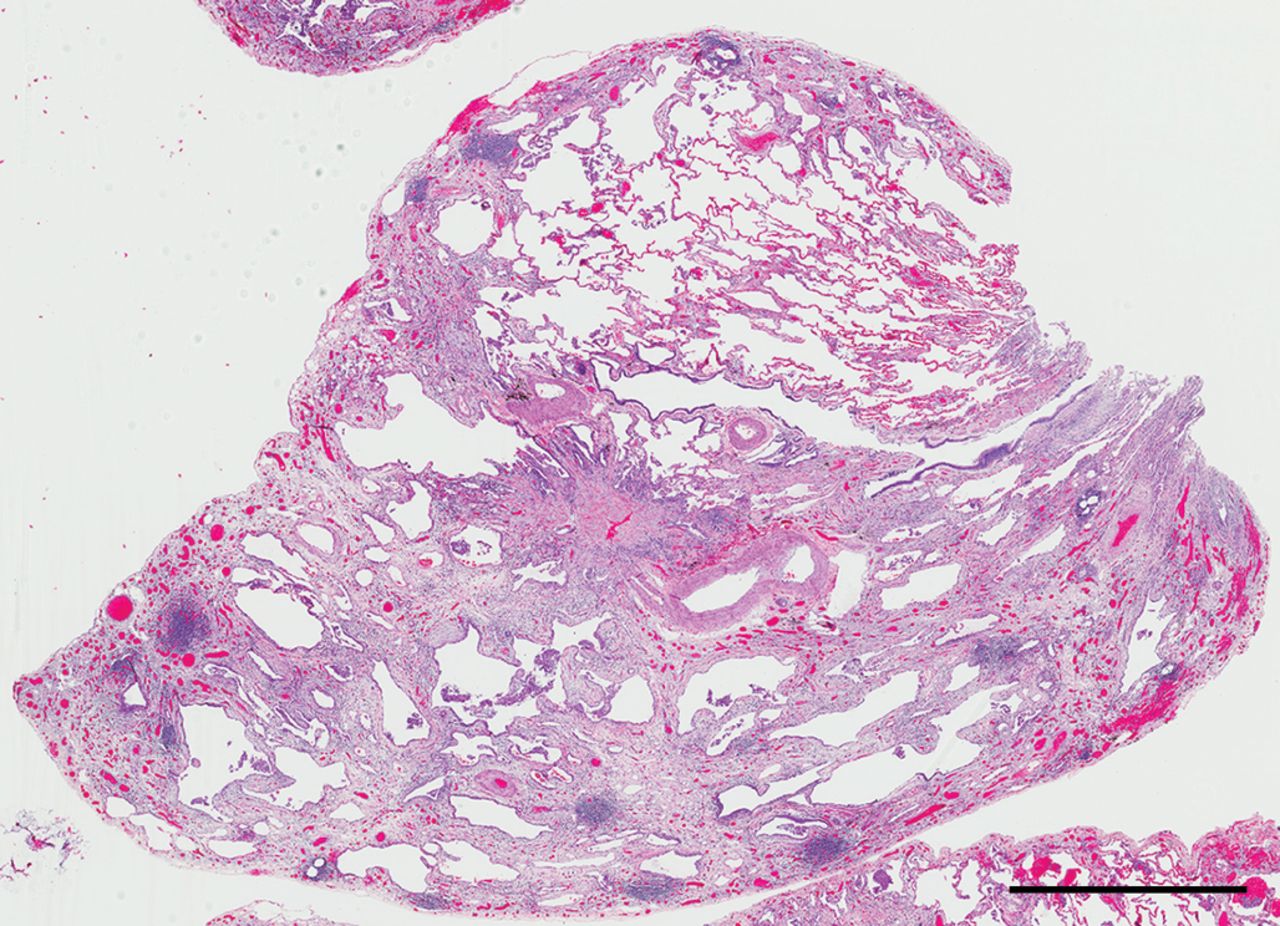
Unclassifiable interstitial fibrosis. This low power view shows patchy fibrosis with subpleural and bronchiolocentric accentuation as well as prominent lymphoid aggregates. Further clinical history for this 68-year-old woman suggested the likely multidisciplinary diagnosis was nitrofurantoin toxicity. Scale bar= 2 mm.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Unclassifiable interstitial fibrosis. This low power view shows patchy fibrosis with focal microscopic honeycombing, airspace enlargement, scattered lymphoid aggregates, and central nonspecific interstitial pneumonia fibrosis-like changes. Further clinical history for this 49-year-old woman with early greying of hair and a sibling with pulmonary fibrosis suggested the likely diagnosis was familial interstitial pulmonary fibrosis. Scale bar=2 mm.
Multidisciplinary diagnosis of patients pathologically unclassifiable by surgical lung biopsy
Post-biopsy evaluation: the MDD
MDD combines the ability of radiology to evaluate the entire lung, the capacity of pathology for investigating features at the cellular level, and the myriad of clinical clues from the interaction of pulmonologist and patient. This combination of data has emerged as a powerful tool in diagnosing interstitial lung disease [51, 52]. The use of the term unclassifiable interstitial lung disease has led to some concern that patients with poorly evaluated disease will be placed into this category without proper due diligence resulting in a group of patients with extraordinary heterogeneity [10]. To address this concern, among others, a working group was assembled to help create a framework or diagnostic ontology for terminology, classification and approach to patients with pulmonary interstitial fibrotic disease [53]. In the proposed schema, patients are divided into four tiers following MDD: those with confident diagnosis (meets current guideline criteria or has >90% certainty), provisional diagnosis with high confidence (70–90% certainty) or low confidence (50–70% certainty), and unclassifiable interstitial lung disease (<50% certainty). The goal of this framework is to help standardise the terminology of fibrotic lung disease. One of the likely consequences of this approach will be a decrease in patients categorised as having unclassifiable interstitial lung disease.
Looking forward
Given the therapeutic differences in managing patients with differing interstitial lung disease, we still need histological evaluation in many cases. However, in current practice, the clinician and radiologist selectively remove the typical cases following recognition of classical patterns on high-resolution computed tomography, presence of environmental antigens, or positive serological testing. The remaining cases are often those with atypical histological features, and the strategies discussed herein become important for recognising potential overlapping pathologies.
Transbronchial cryobiopsy has recently gained attention due to its decreased morbidity when compared with video-assisted thoracoscopic surgery (VATS) biopsy, and increased tissue yields when compared with transbronchial forceps biopsy. These cryobiopsy samples are useful in the diagnosis of the myriad of interstitial lung diseases [54]. However, the amount of tissue is less than that of VATS biopsy, and consequently the percentage of unclassifiable cases increases as expected (1.3% in VATS versus 17.2% in one series) [55]. Given the aforementioned complicated nature of the patients undergoing biopsy, it is difficult to know whether one should promote a technique that hinders the pathologist by the limitation of obtainable tissue [56]. However, there is likely a productive use of this technique in selected patients at experienced centres.
Conclusions
It is the rare person who enjoys not knowing an answer. Physicians are no exception and they like to have information that allows them to make the next therapeutic move. There are actions that can be performed to help decrease the number of unclassifiable cases of interstitial lung disease: using preoperative radiological data to increase the likelihood of obtaining an informative surgical biopsy; performing proper histological processing techniques to maximise evaluable tissue; interpreting patterns of fibrosis and inflammation to construct reasonable differential diagnoses using physiologic principles; and utilising the MDT to incorporate all the clinical, radiological, and histological clues to arrive at a correct diagnosis. In the words of Thomas Henry Huxley, “The known is finite, the unknown infinite; intellectually we stand on an islet in the midst of an illimitable ocean of inexplicability. Our business in every generation is to reclaim a little more land, to add something to the extent and the solidity of our possessions” [57].
Footnotes
Number 6 in the Series “Pathology for the clinician” Edited by Peter Dorfmüller and Alberto Cavazza
Previous articles in this series: No. 1: Ghigna MR, Mooi WJ, Grünberg K. Pulmonary hypertensive vasculopathy in parenchymal lung diseases and/or hypoxia. Eur Respir Rev 2017; 26: 170003. No. 2: Bubendorf L, Lantuejoul S, de Langen AJ, et al. Nonsmall cell lung carcinoma: diagnostic difficulties in small biopsies and cytological specimens. Eur Respir Rev 2017; 26: 170007. No. 3: Rossi G, Cavazza A, Spagnolo P, et al. The role of macrophages in interstitial lung diseases. Eur Respir Rev 2017; 26: 170009. No. 4: Ohshimo S, Guzman J, Costabel U, et al. Differential diagnosis of granulomatous lung disease: clues and pitfalls. Eur Respir Rev 2017; 26: 170012. No. 5: Brandsma C-A, de Vries M, Costa R, et al. Lung ageing and COPD: is there a role for ageing in abnormal tissue repair. Eur Respir Rev 2017; 26: 170073.
Conflict of interest: None declared.
Provenance: Commissioned article, peer reviewed.
- Received December 8, 2017.
- Accepted December 26, 2017.
- Copyright ©ERS 2018.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References











Jump To
- Article
- Abstract
- Abstract
- Introduction
- Surgical lung biopsy: reducing pre-histological barriers to classification
- Typical patterns of fibrosis
- Physiological basis of the typical patterns of fibrosis
- Histological sources of unclassifiable interstitial fibrosis
- Post-biopsy evaluation: the MDD
- Conclusions
- Footnotes
- References
- Figures & Data
- Info & Metrics